Comparative effectiveness of delayed-release dimethyl fumarate versus glatiramer acetate in multiple sclerosis patients: results of a matching-adjusted indirect comparison
Abstract
Aim: Using matching-adjusted indirect comparison, we compared efficacy outcomes in patients with relapsing–remitting multiple sclerosis treated with delayed-release dimethyl fumarate (DMF) or glatiramer acetate (GA). Materials & methods: An indirect comparison of DMF (patient-level data) and GA (aggregate data) was conducted, with average baseline characteristics of DMF patients weighted to match those for GA patients. Direct comparison of DMF and GA was conducted in CONFIRM. Final results pooled the indirect and direct comparisons using meta-analysis. Results: After matching, baseline characteristics were balanced between DMF and GA patients. Compared with GA, efficacy was significantly in favor of DMF as measured by annualized relapse rate (rate ratio: 0.76; 95% CI: 0.57–1.00; p = 0.0474) and 12-week confirmed disability progression (risk ratio: 0.59; 95% CI: 0.46–0.76; p < 0.0001). Conclusion: DMF demonstrated superior clinical efficacy versus GA.
First submitted: 23 November 2016; Accepted for publication: 13 February 2017; Published online: 28 March 2017
Multiple sclerosis (MS) is a chronic, inflammatory, demyelinating disease of the CNS [1] that affects over 400,000 individuals in the USA and 2.5 million worldwide [2]. Lesions caused by the inflammatory process result in symptoms including numbness, fatigue, limb weakness and gait instability, optic neuritis, bowel and bladder dysfunction, cognitive dysfunction, and depression [3,4]. When left untreated, relapses can become more frequent in the active phase of the disease and disability increases over time; these two factors, more frequent relapses and greater disability accumulation, are associated with poorer health-related quality of life [5].
Several disease-modifying therapies (DMTs) have been developed to effectively manage MS symptoms and delay disease progression with minimal adverse effects. Initially, these DMTs were administered parenterally, either via injection (e.g., glatiramer acetate [GA], IFN-β) or infusion (e.g., natalizumab, mitoxantrone), but more recently, DMTs administered orally (e.g., delayed-release dimethyl fumarate [DMF], fingolimod, teriflunomide) have become available [4,6–7]. Although DMTs improve outcomes in patients with MS, there is still a lack of clarity with respect to the benefit–risk of disease compared with treatment. Thus, sometimes therapies with safer profiles may be chosen over those that are perceived to have a greater risk.
DMF is indicated for the treatment of patients with relapsing forms of MS via oral administration and was approved for use in the USA, Canada and Australia in 2013 and for relapsing–remitting MS (RRMS) in the EU in 2014 [8–11]. As of 31 October 2016, over 230,000 patients have been treated with DMF worldwide, representing over 330,000 patient-years of exposure [12]. In Phase III trials (DEFINE and CONFIRM), treatment with DMF 240 mg two times a day (b.i.d.) resulted in significant reductions in annualized relapse rate (ARR), risk of relapse and mean number of new or newly enlarging T2 hyperintense lesions and nonenhancing T1 hypointense lesions compared with placebo (PBO) during the 2-year study periods, and demonstrated favorable benefit–risk profiles in patients with RRMS [13–15]. In an integrated analysis of DEFINE and CONFIRM, the risk of relapse at 2 years was reduced by 43% (p < 0.001) in patients treated with DMF 240 mg b.i.d. (hereafter referred to as DMF) compared with PBO [15].
GA is indicated for the treatment of patients with relapsing forms of MS via subcutaneous injections and was approved for use in the USA in 1996 [16]. In pilot and Phase III trials (US pivotal and European/Canadian pivotal [EUR/CAN]) [17–19], GA treatment resulted in significant relative reductions in clinical disease activity compared with PBO and demonstrated favorable benefit–risk profiles in patients with RRMS. Specifically, there was a significant relative reduction in ARR, mean number of new or newly enlarging T2 hyperintense lesions and nonenhancing T1 hypointense lesions, and improvements in disability (as measured by Expanded Disability Status Scale [EDSS]) in patients treated with GA compared with PBO [17,18]. The mean relapse rate at 2 years was reduced by 29% (p < 0.01) for patients treated with GA compared with PBO [17]. The BEYOND trial found similar efficacy but different adverse event profiles for GA compared with IFN β-1b [20].
Although there is an increasing demand from payers and physicians for comparative effectiveness evaluations to distinguish different treatment options, data from head-to-head trials among these agents are very limited. In the CONFIRM study, GA was included as a reference comparator only, thus the study was not designed or powered for head-to-head comparison of DMF versus GA. Head-to-head comparisons can also be difficult to perform methodologically due to the best practice approach of a double-blinded study design. However, data from multiple trials are available for establishing indirect comparison evidence. Although direct comparisons provide the best evidence, indirect comparisons provide an alternative way to conduct effective comparisons [21]. Matching-adjusted indirect comparisons, unlike other indirect comparison methods, leverage all available data by comparing individual patient data from studies of one agent with summary aggregate data from studies of another agent. The advantage of this method is that it adjusts for the observable aggregate cross-trial differences that might cause potential confounding to the comparison results [21]. However, like all other indirect comparison methods, matching-adjusted indirect comparisons are limited in that they cannot account for confounding resulting from unobservable cross-trial differences [21]. This method has been applied successfully in multiple diseases [22–25]. In the absence of randomized controlled trials involving direct treatment comparisons, indirect treatment comparisons can also be used by healthcare policy makers and payers to assess the decision to prescribe or cover a treatment [26].
GA and DMF are first-line treatments for relapsing forms of MS and are the most likely initial therapies for patients with RRMS, along with fingolimod, teriflunomide and IFN-βs according to recent Association of British Neurologist guidelines [27]. Patients with MS state a preference for oral therapies and indicate oral therapies may improve their quality of life [28]. Patient preference for oral therapies rather than injectable therapies increases as number of treatment switches, and therefore patient experience, grows [29]. Oral therapies may improve DMT adherence given that injection-related reasons, including injection site reactions, injection fatigue and injection anxiety, account for a third of the reasons for nonadherence to injectable DMTs [30]. Compliance was shown to be greater at 6 months in patients taking oral therapies (fingolimod: 77%; teriflunomide: 77%; DMF: 62%) and receiving infusables (natalizumab: 69%) compared with patients receiving injectables (IFN β-1a or -1b or GA: 55%) [31]. Nonadherent patients reported worse quality of life [30].
The objective of this post hoc analysis was to compare the efficacy of DMF with GA on measures of clinical disease activity at 2 years by combining the direct comparison results from CONFIRM and the indirect comparison results from multiple trials using a matching-adjusted indirect approach that adjusted for the observable cross-trial differences.
Materials & methods
Although a head-to-head direct comparison between DMF and GA was underpowered for CONFIRM, it allows for a direct comparison which was used here. For the indirect comparison, patients treated with DMF in the DEFINE and CONFIRM studies were weighted so that after weighting, the weighted mean matched to that of patients treated with GA in the US study, in the EUR/CAN study and in BEYOND (Figure 1 & Table 1).
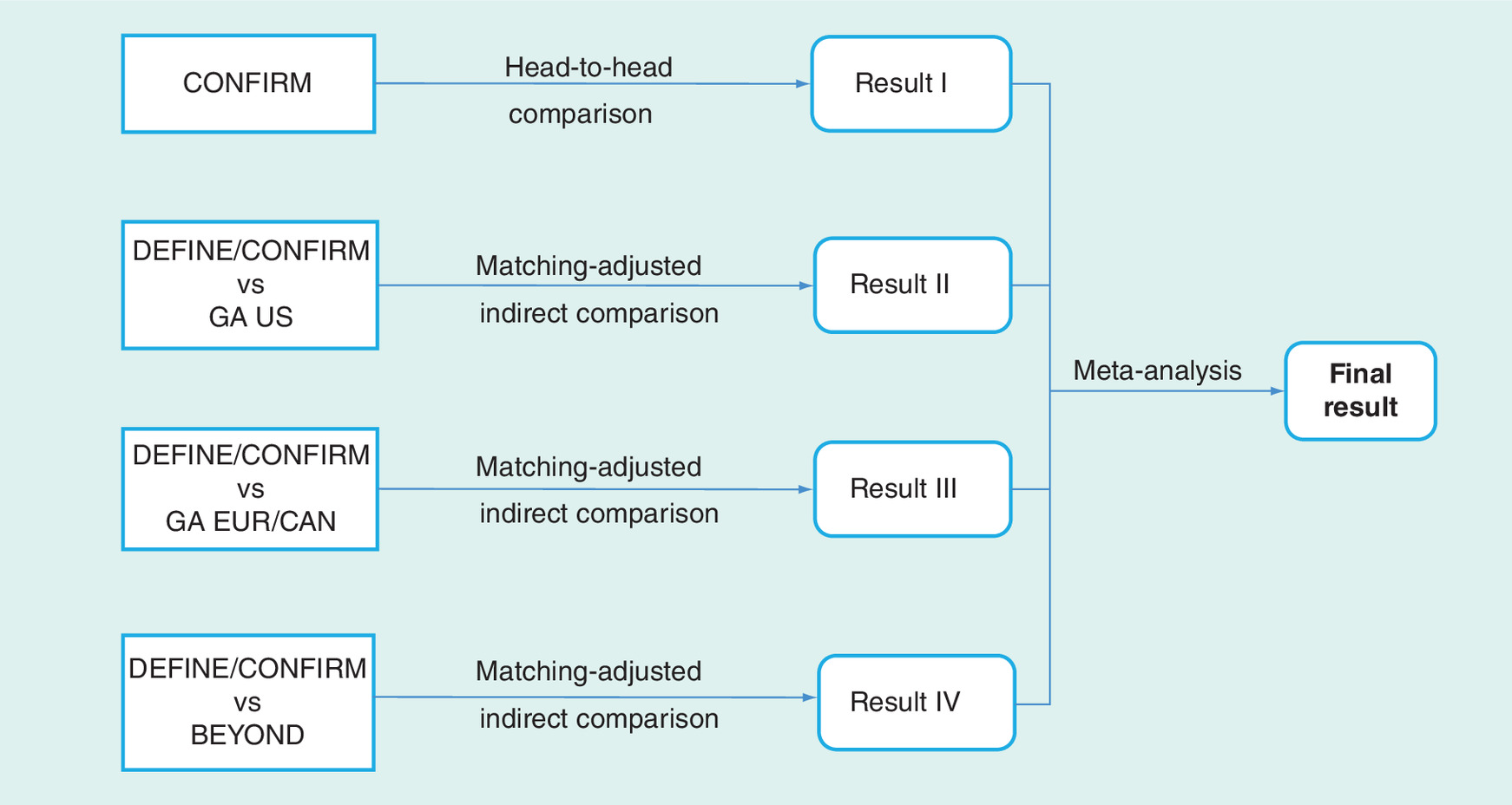
Figure 1. Analysis method.
EUR/CAN: European/Canadian; GA: Glatiramer acetate.
Several studies of GA were not included in this post hoc analysis because the end points were not defined in the same manner as the comparators and therefore the data could not be pooled. Specifically, ARR was either not reported (in Bornstein et al. [19]) or adjusted for the active-reference arm (in REGARD [32], CombiRx [33] and BECOME [34]). All ARR comparisons in this paper were analyzed using ARR adjusted for PBO. ARR adjusted for the active-reference arm could not be used because of the different treatment comparators: REGARD compared GA with IFN β-1a [32], BECOME compared GA with IFN β-1b [34] and CombiRx compared GA with IFN β-1a or treatment with both GA and IFN β-1a [33]. These studies did not compare ARR for GA with PBO. Furthermore, 12-week confirmed disability progression (CDP) was not reported in BECOME [34], REGARD [32], Bornstein et al. [19] or CombiRx [33]. In REGARD [32] and CombiRx [33], 24-week CDP was reported but 12-week CDP was not. Therefore, the following studies were excluded from this analysis: REGARD [32], CombiRx [33], BECOME [34] and Bornstein et al. [19]. Given that the REGARD, CombiRx, BECOME and Bornstein et al. publications did not publish PBO-adjusted ARR or 12-week CDP, we felt that it would not be appropriate or justifiable to include the studies in the primary analysis but we did conduct a sensitivity analysis in which these trials were included. For the sensitivity analysis, first, the DMF arms from DEFINE and CONFIRM were pooled to calculate a pooled mean ARR for DMF; next, the GA arms from CONFIRM, US pivotal, EUR/CAN pivotal, BEYOND, REGARD, CombiRx and Bernstein et al. were pooled to calculate a pooled mean ARR; and finally a ratio of the two pooled mean ARRs for DMF and GA was assessed.
Patients & study design
DMF patient-level data were obtained from two multicenter, double-blind, randomized, Phase III studies: DEFINE, a 2-year, global, PBO-controlled trial (NCT00420212) [14] and CONFIRM, a 2-year, global, PBO-controlled, active-referenced (GA) trial (NCT00451451) [13]. GA aggregate data were obtained from three multicenter, randomized Phase III studies: GA US pivotal, a 2-year, US-based, double-blinded, PBO-controlled trial (NCT00004814) [17]; GA EUR/CAN pivotal, a 9-month, European/Canadian-based, double-blinded, PBO-controlled trial [18]; and BEYOND, a >2-year, global, open-label, active-comparator (IFN β-1b) trial (NCT00099502) [20]. Details of the study designs have been published previously [13–14,17–18,20].
Direct & indirect comparison statistical analysis
For the direct comparison of DMF b.i.d. versus GA in CONFIRM (analysis A), only data from the DMF b.i.d. and GA arms were used. The ARR rate ratios (RR) of DMF versus GA were estimated by a negative binomial model adjusted for baseline EDSS score, age, region and total number of relapses in the 12 months before study entry. A Cox proportional hazards model was used for analysis of time to disability progression adjusted for region, baseline EDSS score and age, where the hazard ratio of DMF versus GA was reported.
For the indirect comparison of DMF versus GA, only data from the DMF b.i.d. in DEFINE/CONFIRM were used and GA data were from other Phase III trials (GA US, GA EUR/CAN and BEYOND; analysis B). PBO data were used across all trials to obtain ARR RRs for active treatment against PBO. Data from the GA arm in CONFIRM was not used in the indirect comparison (analysis B) because such data have already been used in the direct comparison (analysis A) and from a statistical perspective it was preferable not to use them twice. BEYOND employed an active comparator with no PBO arm (Table 1).
Matching-adjusted indirect comparison of DEFINE/CONFIRM and GA trials was conducted as described by Signorovitch et al. [25]. Individual patients in the pooled DMF trials were weighted such that their weighted average baseline characteristics (age, gender, race/ethnicity, time from onset of symptoms, number of relapses in previous year, EDSS [35] and use of any prior DMTs) matched those reported for patients in each GA trial. Of note, the EUR/CAN trial did not provide gender and race/ethnicity data, thus these two variables were not matched for the EUR/CAN trial. Prior treatment was one of the matching criteria, and the GA trials included only treatment-naive patients; thus, when patients from the DMF trials were matched to the GA study populations, only treatment-naive patients remained for comparison.
After matching, a weight was assigned to each individual patient in the DMF trials. With such weights [36], comparisons were made between DMF versus PBO across ARR and 12-week CDP by using the same models as those used in the direct comparison (analysis A) but leveraged by the weights. Of note, comparisons of GA versus PBO were obtained from GA publications. With the two treatments versus PBO comparison results, Bucher’s method was then used to compare DMF versus GA indirectly [36]. Final comparisons of DMF versus GA were obtained by pooling the direct and indirect comparison results of DMF versus GA using random effects meta-analysis. CDP sustained for 24 weeks was not included as an outcome measure in the analysis because most of the GA trials did not assess this end point. The definition of disability progression in DEFINE/CONFIRM was an ≥1.5-point increase from a baseline EDSS score of 0, and an ≥1-point increase from a baseline EDSS score greater than 0. In the GA trials, the definition of disability progression was an ≥1-point increase regardless of baseline value, including patients who had a baseline score of 0.
Sensitivity analysis
A sensitivity analysis was conducted by excluding the BEYOND trial because the BEYOND trial had an active comparator and no PBO arm; all other studies included in this analysis were PBO controlled. Also, an analysis was conducted on the ARR results with and without the GA EUR/CAN trial because the study was only 9 months long.
Results
Patients & trial comparisons
Before matching, the GA-treated patient population was slightly younger and included a greater percentage of white patients than the DMF-treated patient population (Table 2). After matching, baseline characteristics were balanced between DMF and GA patients for each of the matching-adjusted indirect comparisons (Table 3). The effective sample sizes are 1032, 1206 and 364 after matching to the GA US, EUR/CAN, and BEYOND studies, respectively, because all the patients are included but treated with unequal weights. The effective sample size provides the correct sample size for converting the standard deviation of the re-weighted outcome to a standard error. As previously noted, patients in the GA trials did not receive prior DMTs; thus, all patients included in this analysis after matching were DMT naive before entry into the DMF or GA studies.
Comparative effectiveness analysis of DMF versus GA
For the ARR comparison, DEFINE/CONFIRM, GA US and GA EUR/CAN had data available for ARR RR of active treatment versus PBO and were included in the analysis. Before matching, the relative rate reduction for ARR at 2 years favored DEFINE/CONFIRM DMF-treated patients compared with PBO-treated patients (relative rate reduction: 48%; p < 0.0001; Figure 2). Before matching the GA US and EUR/CAN studies, the relative rate reduction for ARR at 2 years was not significantly different between GA-treated patients compared with PBO-treated patients (relative rate reduction: 30%; p = 0.1945 in the GA US study and 33%; p = 0.1504 in the GA EUR/CAN study, Figure 2). After matching to GA US and EUR/CAN studies, the relative rate reduction for ARR at 2 years favored DEFINE/CONFIRM DMF-treated patients compared with PBO-treated patients (relative rate reduction: 46%; p < 0.0001 and 55%; p < 0.0001, respectively). Compared with GA, efficacy was significantly in favor of DMF as measured by the ARR with GA as the reference (relative rate reduction: 24%; p = 0.0474; Figure 2).
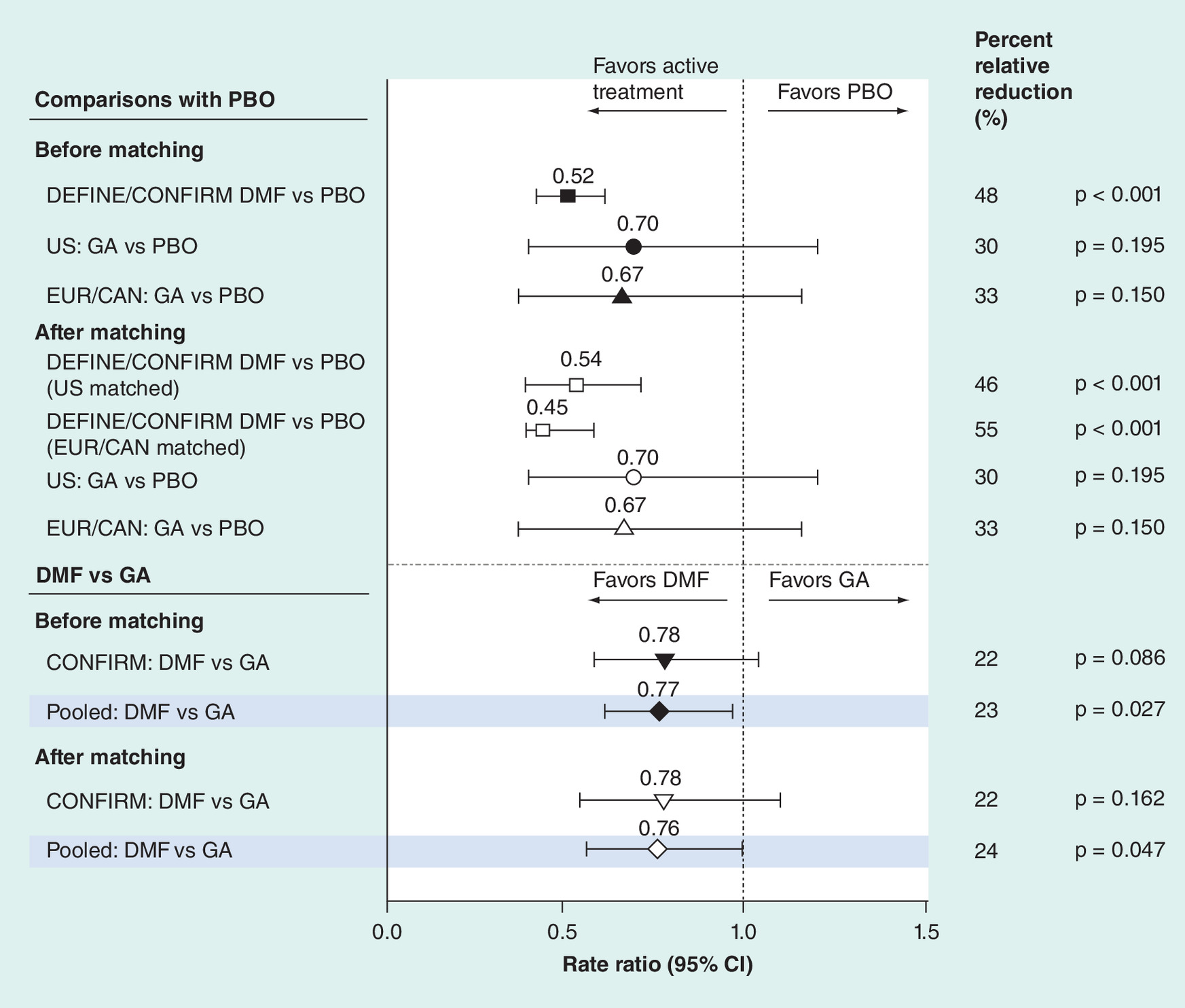
Figure 2. Annualized relapse rate at 2 years†.
†Rate ratio <1 indicates an improved outcome for DMF relative to GA, DMF relative to PBO or GA relative to PBO.
DMF: also known as gastro-resistant DMF.
Data not shown for the DEFINE/CONFIM versus BEYOND comparison because ARR adjusted for PBO was not available for BEYOND. The standard error of the rate ratio of GA versus PBO used for p-value calculation was approximated by:
Standard error of DMF vs PBO × Square root (sample size of GA trial) Square root (sample size of DMF trial). Closed symbols indicate risk ratio before matching, open symbols indicate risk ratio after matching, and shaded lines represent risk ratios from the pooled data.
DMF: Delayed-release dimethyl fumarate; EUR/CAN: European/Canadian; GA: Glatiramer acetate; PBO: Placebo.
For efficacy measured by 12-week CDP, the relative risk reduction favored DEFINE/CONFIRM DMF-treated patients compared with PBO-treated patients before matching (relative risk reduction: 34%; p < 0.001) as well as after (relative risk reduction: 46%; p < 0.002; Figure 3). For efficacy measured by 12-week CDP, the relative risk reduction was not significantly different between GA-treated patients and PBO-treated patients in the GA US study before matching (relative risk reduction: 12%; p = 0.5734; Figure 3). Compared with GA, efficacy was significantly in favor of DMF as measured by 12-week CDP (relative risk reduction: 41%; p < 0.0001) with GA as the reference (Figure 3).
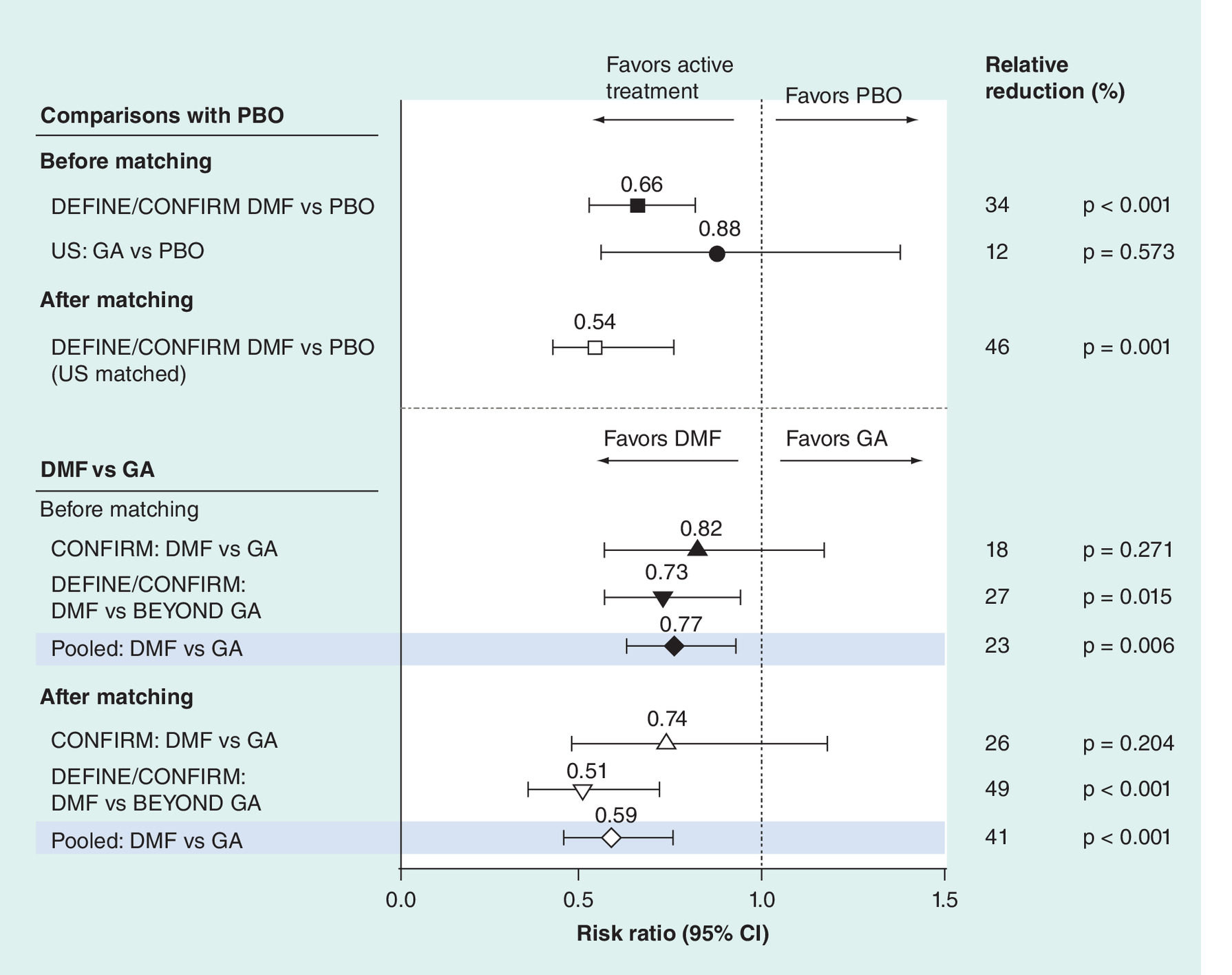
Figure 3. 12-week confirmed disability progression†.
†Risk ratio <1 indicates an improved outcome for DMF relative to GA, DMF relative to PBO or GA relative to PBO.
DMF: also known as gastro-resistant DMF.
Confirmed disability progression in GA US and BEYOND was defined as an ≥1-point increase in EDSS score sustained for 12 weeks. Data not shown for the GA EUR/CAN versus PBO comparison because CDP data were not available for GA EUR/CAN. p-value of the risk ratio for DMF versus PBO and GA versus PBO was based on the chi-square test. Closed symbols indicate risk ratio before matching, open symbols indicate risk ratio after matching and shaded lines represent risk ratios from the pooled data.
DMF: Delayed-release dimathyl fumarate; EDSS: Expanded Disability Status Scale; GA: Glatiramer acetate; PBO: Placebo.
Sensitivity analysis
In BEYOND, there was an active comparator and no PBO arm, and therefore a sensitivity analysis was done in which arm-to-arm indirect comparison result of DEFINE/CONFIRM versus BEYOND was excluded. In this sensitivity analysis, the risk ratio was 0.69 (95% CI: 0.48–0.98; p = 0.041), consistent with those obtained in the primary analysis. The effect of the shorter study period for GA EUR/CAN on ARR was assessed using a sensitivity analysis. The trend of effect sizes is quite similar with or without the trial.
Discussion
Because data from head-to-head trials are limited, indirect comparisons remain important, albeit imperfect, tools to address the comparative effectiveness of drugs. In order to further assess the relative effect of DMF versus GA, we performed a matching-adjusted indirect comparison of DMF with GA. Using this method, DMF demonstrated significantly favorable clinical efficacy compared with GA on ARR and also on 12-week CDP.
Other studies have compared DMTs for MS using different methods and obtained comparable results. A systematic review and data synthesis of DMF and other DMTs using mixed treatment comparisons found DMF significantly reduced ARR compared with GA (RR: 0.795 [95% CI: 0.668–0.947]) [37]. In an adjusted indirect comparison of DMF, teriflunomide and laquinimod, using Bucher’s adjusted indirect comparison method, it was found that relapse risk was lower with DMF compared with laquinimod or teriflunomide [38]. Roskell et al. performed an indirect comparison of ARR using mixed treatment comparison framework for fingolimod versus GA, IFN β-1b and IFN β-1a [39]. Fingolimod significantly reduced relapse frequency compared with injectable first-line DMTs. Zagmutt et al. performed a meta-analysis comparing adverse events for DMF, GA and teriflunomide, and showed that patients treated with GA may have better quality of life compared with DMF or teriflunomide [40]. A recent indirect comparison of DMF versus fingolimod, using the same matching-adjusted comparison method, found DMF and fingolimod had similar effects on ARR, 12-week CDP, no evidence of disease activity and the Multiple Sclerosis Functional Composite, while DMF demonstrated greater benefit on the EuroQoL 5-Dimensions questionnaire utility score and visual analog scale compared with fingolimod [41].A recent efficacy comparison using registry data found similar results. This comparative analysis of efficacy between DMF, fingolimod, GA, teriflunomide and IFN compared data from the MSBase registry using binomial propensity score matching followed by a generalized estimating Poisson model offset to estimate treatment exposure for ARRs [42]. The ARR was slightly higher in the DMF group compared with the matched fingolimod group (0.22 vs 0.19), while ARR was lower in the DMF group relative to either GA (0.24 vs 0.26), teriflunomide (0.17 vs 0.27) or IFN (0.23 vs 0.26) [42]. The methods used in the indirect comparison presented here are different to those used in the comparisons described above, yet similar results were found. Although it is possible that using alternate methods to conduct this comparison would lead to different results due to different assumptions made in the model, we felt the matching-adjusted indirect comparison described by Signorovitch [25] and used here is the most appropriate, based on the available data.
Health economics studies have also used similar methods. Economic models using acquisition and healthcare cost data from Canada, France and the USA have shown that DMF is cost-effective compared with GA and fingolimod [43–46] Dorman et al. found the budget impact of adding DMF as a treatment would be partially offset by the reduced costs of relapses [47].
Smaller studies or small subsets of patients within studies allow direct comparisons of DMF and GA [13]. In CONFIRM, GA was included as reference arm. For both DMF and GA compared with PBO, a reduced rate of relapse, reduced mean number of new T1 hypointense lesions, greater percentage of patients free from new or newly enlarging T2 hyperintense lesions and decreased odds of more gadolinium-enhancing lesions at 2 years were observed [13]. However, the study was not significantly powered to compare the DMF and GA arms [13]. A post hoc analysis of this subset of patients in CONFIRM found inflammatory disease activity over 2 years was significantly reduced in patients treated with DMF compared with patients treated with GA [48].
The time period of the studies included in this comparison is an important factor given changes in MS diagnosis, MS patient care, including timing of DMT treatment initiation, and known reductions in ARR that occurred over the first decade of the DMT era. Because the early pivotal trials had higher relapse rates, RRs were used instead of absolute rate for the ARR analysis. By using ARR RR, we found that the comparison results of DMF compared with GA are similar across different GA studies conducted in different years. That is, the RR of DMF compared with GA in CONFIRM (RR: 0.78 for both before and after matching), which was conducted in 2012, is quite similar to the RR of DMF compared with GA in the US pivotal study, which was conducted in 1990 (RR: 0.73 before matching and 0.76 after matching) and EUR/CAN in approximately 2000 (RR: 0.77 before matching and 0.67 after matching). The typical time delay between DMT treatment initiation relative to MS diagnosis had changed since some of the trials used in this analysis occurred. These potential timing differences to a certain point were reflected in the differences of patient demographics and MS disease characteristics and served as a rationale for the matching adjustment performed in this analysis. The more recent studies (CONFIRM, DEFINE and BEYOND) have shown similar results that corroborate the results of the indirect comparison involving the two old GA trials and as a result, relieve the author’s concern of bias due to timing difference.
Although providing a valuable indirect comparison, the present analysis is not without limitation. This analysis relied on indirect comparison evidence, which involves all the caveats and limitations of an observational study, including confounding, selection and definitions of end points, and data availability [49]. Inclusion criteria were similar across trials however, differences are worth noting. Age varied between trials, with younger patients in the GA trials. Specifically, patients were aged 18–45 years in the GA US trial, 18–50 years in the GA EUR/CAN trial, and 18–55 years in DEFINE/CONFIRM and BEYOND. Also, the trials used different versions of the McDonald diagnostic criteria [50]. The definition of disability progression was inconsistent across trials and there may have been differences in how missing EDSS scores were handled and what was done if a relapse occurred together with EDSS progression, which may have led to differences or bias across trials. Also, patients in GA trials were naive to prior DMTs. Matching can account for the difference of age and proportions of DMT-naive patients; therefore, those factors were matched in the matching-adjusted indirect comparison. It was not possible to match the others, so residual confounders and bias due to the differences discussed above may have impacted the results [25]. In addition, the 2-year duration may not be sufficient to detect significance with CDP and also may not be long enough to assess the durability of the ARR treatment effect. The differences in defining CDP between trials may also limit the conclusions. The potential effects of unobserved or unknown differences are of course unknown. Finally, some trials were excluded because the end points were not defined in the same manner as the comparators and therefore the data could not be pooled. A sensitivity analysis was performed using a simple comparison as described in the 'Materials & methods' section. The results showed that the ARR relative rate reduction of DMF versus GA was 34% for adjusted ARR and 46% for unadjusted ARR. Although there are many caveats to this approach, these results are consistent with the more statistically rigorous matching-adjusted indirect comparison (24%) presented in this manuscript and suggest that the exclusion of the studies dampens rather than amplifies the percent relative reduction calculated for DMF compared with GA.
Conclusion
DMF demonstrated significantly favorable clinical efficacy for both ARR and 12-week CDP versus GA in this matching-adjusted indirect comparison.
| Study | Arms | Study duration (year) | Primary end point | ARR | 12-week CDP |
|---|---|---|---|---|---|
| DEFINE | DMF vs PBO | 2 | Time to relapse | Yes | Yes |
| CONFIRM | DMF vs GA vs PBO | 2 | Time to relapse | Yes | Yes |
| GA US | GA vs PBO | 2 | ARR | Yes | Yes |
| GA EUR/CAN | GA vs PBO | 1 | Gd lesions | Yes | No |
| BEYOND | GA vs IFN β-1b | 2 | Time to relapse | No | Yes |
ARR data adjusted for PBO are not available for BEYOND.
ARR: Annual relapse rate; CDP: Confirmed disability progression; DMF: Delayed-release dimethyl fumarate; GA: Glatiramer acetate; Gd: Gadolinium-enhancing; IFN: Interferon; PBO: Placebo.
| Characteristics | Before matching | ||||
|---|---|---|---|---|---|
| DMF † | GA | ||||
| DEFINE | CONFIRM | US study | EUR/CAN study | BEYOND | |
| n | 1234 | 1417 | 251 | 239 | 448 |
| Age (years) | 38.5 | 37.3 | 34.5 | 34.1 | 35.2 |
| Female (%) | 73.6 | 70.0 | 73.3 | NR | 68 |
| White (%) | 79 | 84 | 94 | NR | 91 |
| Duration of MS (years) | 8.3 | 7.7 | 7.0 | 8.1 | 5.1 |
| Prior 1-year relapse rate | 1.3 | 1.4 | 1.5‡ | 1.3‡ | 1.6 |
| EDSS score | 2.4 | 2.6 | 2.6 | 2.4 | 2.3 |
| Any prior DMT (%)§ | 41 | 29 | 0 | 0 | 0 |
†DMF: also known as gastro-resistant DMF.
‡The prior 1-year relapse rate is approximated by half of the prior 2-year relapse rate.
§The three GA trials do not allow pre-DMT-treated patients.
DMF: Delayed-release dimethyl fumarate; DMT: Disease-modifying therapy; EDSS: Expanded Disability Status Scale; GA: Glatiramer acetate; MS: Multiple sclerosis; NR: Not reported.
| Characteristics | DEFINE/CONFIRM DMF† | ||
|---|---|---|---|
| US matched | EUR/CAN matched | BEYOND matched | |
| n‡ | 1032 | 1206 | 364 |
| Age (years) | 34.5 | 34.1 | 35.2 |
| Female (%) | 73.3 | NR | 68 |
| White (%) | 94 | NR | 91 |
| Duration of MS (years) | 7.0 | 8.1 | 5.1 |
| Prior 1-year relapse rate | 1.5§ | 1.3§ | 1.6 |
| EDSS score | 2.6 | 2.4 | 2.3 |
| Any prior DMT (%)§ | 0 | 0 | 0 |
†DMF: also known as gastro-resistant DMF.
‡n is the effective sample size that is computed as the square of the summed weights divided by the sum of the squared weights.
§The prior 1-year relapse rate is approximated by half of the prior 2-year relapse rate.
DMF: Delayed-release dimethyl fumarate; DMT: Disease-modifying therapy; EDSS: Expanded Disability Status Scale; GA: Glatiramer acetate; EUR/CAN: European/Canadian; MS: multiple sclerosis; NR: Not reported.
Matching-adjusted indirect comparisons can be useful when head-to-head comparisons are not available.
This was a matching-adjusted indirect comparison between delayed-release dimethyl fumarate (DMF) and glatiramer acetate (GA), two first-line treatments for relapsing forms of multiple sclerosis (MS).
Patients in the GA trials were DMT-naive; therefore, only DMT-naive patients from the DMF trials were included. Efficacy, as measured by annualized relapse rate, was significantly in favor of DMF, with GA as the reference (relative rate reduction: 24%; p = 0.0474).
For 12-week confirmed disability progression, the relative risk reduction was 41% (p < 0.0001) in favor of DMF, with GA as the reference.
A sensitivity analysis assessing the effect of a shorter study period showed similar results.
Overall, DMF demonstrated significantly favorable clinical efficacy for both annualized relapse rate and 12-week confirmed disability progression versus GA in this matching-adjusted indirect comparison.
Acknowledgements
These data were previously presented as a poster at the second Congress of the European Academy of Neurology, Copenhagen, Denmark, 28–31 May 2016.
Financial & competing interests disclosure
A Chan: compensation for activities with Allmirall Hermal, Bayer, Biogen, Genzyme, Merck, Novartis, Roche, Sanofi -Aventis and Teva Neuroscience; research support from Biogen, Genzyme, and Novartis. G Cutter: serves on data and safety monitoring committees for Apotek, Biogen, Cleveland Clinic (Vivus), GlaxoSmithKline Pharmaceuticals, Gilead Pharmaceuticals, Modigenetech/Prolor, Merck/Ono Pharmaceuticals, Merck, Merck/Pfizer, Neuren, Sanofi-Aventis, Teva, NHLBI (Protocol Review Committee), NINDS, and NICHD (OPRU oversight committee); and receives consulting or speaking fees from Consortium of MS Centers (grant), D3 (Drug Discovery and Development), Genzyme, Jannsen Pharmaceuticals, Klein-Buendel Incorporated, Medimmune, Novartis, Opexa Therapeutics, Receptos, Roche, EMD Serono, Teva Pharmaceuticals, Transparency Life Sciences. RJ Fox: consultant fees from Biogen, MedDay, Novartis, Questcor, Teva, and Xenoport; advisory committees for Biogen, and Novartis; research grant funding from Novartis. J Xiao, JB Lewin and MR Edwards: employees of and hold stock/stock options in Biogen. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Biogen provided funding for medical writing support in the development of this paper; Karen Spach, PhD, CMPP, from Excel Scientific Solutions wrote the first draft of the manuscript based on input from authors, and Kristen DeYoung from Excel Scientific Solutions copyedited and styled the manuscript per journal requirements. Biogen reviewed and provided feedback on the paper to the authors.
Ethical conduct of research
The authors state that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations. In addition, for investigations involving human subjects, informed consent has been obtained from the participants involved.
Open access
This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
1.
Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. N. Engl. J. Med. 343(13), 938–952 (2000).
2.
Tullman MJ. Overview of the epidemiology, diagnosis, and disease progression associated with multiple sclerosis. Am. J. Manage. Care 19(Suppl. 2), S15–S20 (2013).
3.
Compston A, Coles A. Multiple sclerosis. Lancet 372(9648), 1502–1517 (2008).
4.
Deeks ED. Dimethyl fumarate: a review in relapsing–remitting MS. Drugs 76(2), 243–254 (2016).
5.
Turpin RS, Blumberg PB, Sharda CE, Salvucci LA, Haggert B, Simmons JB. Patient adherence: present state and future directions. Dis. Manag. 10(6), 305–310 (2007).
6.
Fox RJ, Kita M, Cohan SL et al. BG-12 (dimethyl fumarate): a review of mechanism of action, efficacy, and safety. Curr. Med. Res. Opin. 30(2), 251–262 (2014).
7.
Kretzschmar B, Pellkofer H, Weber MS. The use of oral disease-modifying therapies in multiple sclerosis. Curr. Neurol. Neurosci. Rep. 16(4), 38 (2016).
8.
Biogen Inc. Tecfidera (dimethyl fumarate) delayed-release capsules: US prescribing Information. 2016 (May 31) (2014). www.accessdata.fda.gov/drugsatfda_docs/appletter/2013/204063Orig1s000ltr.pdf
9.
Biogen Inc. Product Information for AusPAR Tecfidera Dimethyl Fumarate. 2016 (July 18) (2013). www.accessdata.fda.gov/drugsatfda_docs/nda/2014/204063Orig1s010.pdf
10.
Biogen Idec Ltd. Tecfidera gastro-resistant hard capsules: EU summary of product characteristics. 2016 (May 31) (2015). www.medicines.org.uk/emc/medicine/28593
11.
Biogen Inc. TecfideraTM dimethyl fumarate delayed-release capsules: Canadian product monograph. (2015). www.biogen.ca/content/dam/corporate/en_CA/pdfs/products/TECFIDERA/1-TECFIDERA-PM-2015-01-29-E.pdf
12.
Biogen. Tecfidera Exposure Letter for Data through 31 Oct 2016 (2016).
13.
Fox RJ, Miller DH, Phillips JT et al. Placebo-controlled Phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N. Engl. J. Med. 367(12), 1087–1097 (2012).
•• This is a pivotal study included in the indirect comparison.
14.
Gold R, Kappos L, Arnold DL et al. Placebo-controlled Phase 3 study of oral BG-12 for relapsing multiple sclerosis. N. Engl. J. Med. 367(12), 1098–1107 (2012).
•• This is a pivotal study included in the indirect comparison.
15.
Viglietta V, Miller D, Bar-Or A et al. Efficacy of delayed-release dimethyl fumarate in relapsing–remitting multiple sclerosis: integrated analysis of the Phase III trials. Ann. Clin. Trans. Neurol. 2(2), 103–118 (2015).
16.
Teva Neuroscience I. COPAXONE (glatiramer acetate injection) for subcutaneous use: US prescribing Information. (2016). www.copaxone.com/resources/pdfs/prescribinginformation.pdf
17.
Johnson KP, Brooks BR, Cohen JA The Copolymer 1 Multiple Sclerosis Study Group Copolymer 1 reduces relapse rate and improves disability in relapsing–remitting multiple sclerosis: results of a Phase III multicenter, double-blind placebo-controlled trial. Neurology 45(7), 1268–1276 (1995).
•• This is a pivotal study included in the indirect comparison.
18.
Comi G, Filippi M, Wolinsky JS. European/Canadian Glatiramer Acetate Study Group European/Canadian multicenter, double-blind, randomized, placebo-controlled study of the effects of glatiramer acetate on magnetic resonance imaging-measured disease activity and burden in patients with relapsing multiple sclerosis. Ann. Neurol. 49(3), 290–297 (2001).
•• This is a pivotal study included in the indirect comparison.
19.
Bornstein MB, Miller A, Slagle S et al. A pilot trial of Cop 1 in exacerbating-remitting multiple sclerosis. N. Engl. J. Med. 317(7), 408–414 (1987).
20.
O’Connor P, Filippi M, Arnason B et al. 250 microg or 500 microg interferon beta-1b versus 20 mg glatiramer acetate in relapsing–remitting multiple sclerosis: a prospective, randomised, multicentre study. Lancet Neurol. 8(10), 889–897 (2009).
•• This is a pivotal study included in the indirect comparison.
21.
Signorovitch JE, Sikirica V, Erder MH et al. Matching-adjusted indirect comparisons: a new tool for timely comparative effectiveness research. Value Health 15(6), 940–947 (2012).
22.
Signorovitch JE, Wu EQ, Betts KA et al. Comparative efficacy of nilotinib and dasatinib in newly diagnosed chronic myeloid leukemia: a matching-adjusted indirect comparison of randomized trials. Curr. Med. Res. Opin. 27(6), 1263–1271 (2011).
23.
Signorovitch JE, Wu EQ, Swallow E, Kantor E, Fan L, Gruenberger JB. Comparative efficacy of vildagliptin and sitagliptin in Japanese patients with Type 2 diabetes mellitus: a matching-adjusted indirect comparison of randomized trials. Clin. Drug Invest. 31(9), 665–674 (2011).
24.
Signorovitch J, Erder MH, Xie J et al. Comparative effectiveness research using matching-adjusted indirect comparison: an application to treatment with guanfacine extended release or atomoxetine in children with attention-deficit/hyperactivity disorder and comorbid oppositional defiant disorder. Pharmacoepidemiol. Drug Saf. 21(Suppl. 2), 130–137 (2012).
25.
Signorovitch JE, Wu EQ, Yu AP et al. Comparative effectiveness without head-to-head trials: a method for matching-adjusted indirect comparisons applied to psoriasis treatment with adalimumab or etanercept. Pharmacoeconomics 28(10), 935–945 (2010).
• Details the statistical methods used in this manuscript.
26.
Jansen JP, Fleurence R, Devine B et al. Interpreting indirect treatment comparisons and network meta-analysis for health-care decision making: report of the ISPOR Task Force on Indirect Treatment Comparisons Good Research Practices: part 1. Value Health 14(4), 417–428 (2011).
27.
Scolding N, Barnes D, Cader S et al. Association of British Neurologists: revised (2015) guidelines for prescribing disease-modifying treatments in multiple sclerosis. Prac. Neurol. 15(4), 273–279 (2015).
28.
Canadian Agency for Drugs and Technologies in Health. CADTH therapeutic review: recommendations for drug therapies for relapsing–remitting multiple sclerosis (2013). www.cadth.ca/media/pdf/TR0004_RRMS_RecsReport_TR_e.pdf
29.
Morawski J HS, Cox F, Turkistani F. Treatment preferences related to route of administration in patients with MS: results from US and EU5. Mult. Scler. 22(Suppl. 3), 807–808 (2016).
30.
Devonshire V, Lapierre Y, Macdonell R et al. The Global Adherence Project (GAP): a multicenter observational study on adherence to disease-modifying therapies in patients with relapsing–remitting multiple sclerosis. Eur. J. Neurol. 18(1), 69–77 (2011).
31.
Duquette P, Rivest D, Selchen D et al. A retrospective claims analysis: compliance and discontinuation rates among Canadian MS patients treated with disease-modifying therapies - Canadian real world experience. Mult. Scler. 22(Suppl. 3), 149–150, P374 (2016).
32.
Mikol DD, Barkhof F, Chang P et al. Comparison of subcutaneous interferon beta-1a with glatiramer acetate in patients with relapsing multiple sclerosis (the REbif vs Glatiramer Acetate in Relapsing MS Disease [REGARD] study): a multicentre, randomised, parallel, open-label trial. Lancet Neurol. 7(10), 903–914 (2008).
33.
Lublin FD, Cofield SS, Cutter GR et al. Randomized study combining interferon and glatiramer acetate in multiple sclerosis. Ann. Neurol. 73(3), 327–340 (2013).
34.
Cadavid D, Wolansky LJ, Skurnick J et al. Efficacy of treatment of MS with IFNbeta-1b or glatiramer acetate by monthly brain MRI in the BECOME study. Neurology 72(23), 1976–1983 (2009).
35.
Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology 33(11), 1444–1452 (1983).
36.
Bucher HC, Guyatt GH, Griffith LE, Walter SD. The results of direct and indirect treatment comparisons in meta-analysis of randomized controlled trials. J. Clin. Epidemiol. 50(6), 683–691 (1997).
37.
Hutchinson M, Fox RJ, Havrdova E et al. Efficacy and safety of BG-12 (dimethyl fumarate) and other disease-modifying therapies for the treatment of relapsing–remitting multiple sclerosis: a systematic review and mixed treatment comparison. Curr. Med. Res. Opin. 30(4), 613–627 (2014).
• A similar indirect comparison of dimethyl fumarate and other disease-modifying treatments are presented in this publication.
38.
Metin H, Huppertz H. Adjusted indirect comparison of oral multiple sclerosis agents. Value Health 18(7), A750 (2015).
39.
Roskell NS, Zimovetz EA, Rycroft CE, Eckert BJ, Tyas DA. Annualized relapse rate of first-line treatments for multiple sclerosis: a meta-analysis, including indirect comparisons versus fingolimod. Curr. Med. Res. Opin. 28(5), 767–780 (2012).
40.
Zagmutt FJ, Carroll CA. Meta-analysis of adverse events in recent randomized clinical trials for dimethyl fumarate, glatiramer acetate and teriflunomide for the treatment of relapsing forms of multiple sclerosis. Int. J. Neurosci. 125(11), 798–807 (2015).
41.
Fox RJ, Chan A, Zhang A et al. Comparative effactiveness using amatching-adjusted indirect comparison betwen delayed-release dimethyl fumarateand fingoimod for the treatment of multiple sclerosis. Curr. Med. Res. Opin. 33(2), 175–183 (2016).
42.
Spelman T, Kalincik T, Trojano M et al. Comparative analysis of MS outcomes in dimethyl fumarate-treated patients relative to propensity matched fingolimod, teriflunomide, interferon or glatiramer acetate. Presented at: 32nd Congress of the European Committee for Treatment & Research in Multiple Sclerosis. London, UK, 14–17 September 2016 (Abstract P1157).
43.
Mauskopf J, Fay M, Iyer R, Sarda S, Livingston T. Cost-effectiveness of delayed-release dimethyl fumarate for the treatment of relapsing forms of multiple sclerosis in the United States. J. Med. Econ. 19(4), 432–442 (2016).
44.
Chevalier J, Chamoux C, Hammes F, Chicoye A. Cost-effectiveness of treatments for relapsing remitting multiple sclerosis: a French societal perspective. PLoS ONE 11(3), e0150703 (2016).
45.
Mauskopf J FM, Iyer R, Sarda S, Livingston T. Cost-effectiveness of delayed-release dimethyl fumarate compared with glatiramer acetate and fingolimod for the treatment of relapsing–remitting multiple sclerosis. Value Health 17(3), A60–A61 (2014).
46.
Su W, Kansal A, Vicente C, Deniz B, Sarda S. The cost-effectiveness of delayed-release dimethyl fumarate for the treatment of relapsing–remitting multiple sclerosis in Canada. J. Med. Econ. 9(7), 718–727 (2016).
47.
Dorman E, Kansal AR, Sarda S. The budget impact of introducing delayed-release dimethyl fumarate for treatment of relapse-remitting multiple sclerosis in Canada. J. Med. Econ. 18(12), 1085–1091 (2015).
48.
Kremenchutzky M, Fox Rj, Phillips Jt et al. Efficacy of delayed-release dimethyl fumarate vs glatiramer acetate on a novel composite outcome measure of inflammatory disease activity: post-hoc analysis of the CONFIRM study. Mult. Scler. 23(S11), 545–546 (2015).
49.
Carlson MD, Morrison RS. Study design, precision, and validity in observational studies. J. Palliative Med. 12(1), 77–82 (2009).
50.
Polman CH, Reingold SC, Banwell B et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann. Neurol. 69(2), 292–302 (2011).
Information & Authors
Information
Published In
Copyright
© Biogen.
History
Published online: 28 March 2017
Keywords:
Topics
Authors
Metrics & Citations
Metrics
Article Usage
Article usage data only available from February 2023. Historical article usage data, showing the number of article downloads, is available upon request.
Citations
How to Cite
Comparative effectiveness of delayed-release dimethyl fumarate versus glatiramer acetate in multiple sclerosis patients: results of a matching-adjusted indirect comparison. (2017) Journal of Comparative Effectiveness Research. DOI: 10.2217/cer-2016-0085
Export citation
Select the citation format you wish to export for this article or chapter.
Citing Literature
- Mostafa A. Rabie, Henning Madry, Magali Cucchiarini, Nesrine S. El-Sayed, The brain-joint axis: links between osteoarthritis and neurodegenerative disorders in aging, Journal of Advanced Research, 10.1016/j.jare.2025.10.023, 85, (1119-1152), (2026).
- Owen Cassidy, Marie Harte, Lea Trela-Larsen, Cathal Walsh, Arthur White, Laura McCullagh, Joy Leahy, A Comparison of Relative-Efficacy Estimate(S) Derived From Both Matching-Adjusted Indirect Comparisons and Standard Anchored Indirect Treatment Comparisons: A Review of Matching-Adjusted Indirect Comparisons, Value in Health, 10.1016/j.jval.2023.07.001, 26, 11, (1665-1674), (2023).
- Ralf Gold, Michael Barnett, Andrew Chan, Huiyu Feng, Kazuo Fujihara, Gavin Giovannoni, Xavier Montalbán, Fu-Dong Shi, Mar Tintoré, Qun Xue, Chunsheng Yang, Hongyu Zhou, Clinical use of dimethyl fumarate in multiple sclerosis treatment: an update to include China, using a modified Delphi method, Therapeutic Advances in Neurological Disorders, 10.1177/17562864231180734, 16, (2023).
- Tammy Jiang, Tjalf Ziemssen, Sibyl Wray, Changyu Shen, Karin Söderbärg, James B. Lewin, Ivan Božin, Mark S. Freedman, Matching-Adjusted Indirect Comparisons of Diroximel Fumarate, Ponesimod, and Teriflunomide for Relapsing Multiple Sclerosis, CNS Drugs, 10.1007/s40263-023-01002-x, 37, 5, (441-452), (2023).
- Moisés Manuel Gallardo Pérez, Solón Javier Garcés Eisele, MicroRNAs as a possible biomarker in the treatment of multiple sclerosis, IBRO Neuroscience Reports, 10.1016/j.ibneur.2022.11.001, 13, (492-499), (2022).
- Yoko Yagishita, Tonibelle N. Gatbonton-Schwager, Melissa L. McCallum, Thomas W. Kensler, Current Landscape of NRF2 Biomarkers in Clinical Trials, Antioxidants, 10.3390/antiox9080716, 9, 8, (716), (2020).
- Jashin J. Wu, Jes B. Hansen, Dharm S. Patel, Nanna Nyholm, Karen A. Veverka, Andrine R. Swensen, Effectiveness comparison and incremental cost-per-responder analysis of calcipotriene 0.005%/betamethasone dipropionate 0.064% foam vs. halobetasol 0.01%/tazarotene 0.045% lotion for plaque psoriasis: a matching-adjusted indirect comparative analysis, Journal of Medical Economics, 10.1080/13696998.2020.1722139, 23, 6, (641-649), (2020).
- Mikah S. Brandes, Nora E. Gray, NRF2 as a Therapeutic Target in Neurodegenerative Diseases, ASN Neuro, 10.1177/1759091419899782, 12, 1, (2020).
- Hannah A. Blair, Dimethyl Fumarate: A Review in Relapsing-Remitting MS, Drugs, 10.1007/s40265-019-01229-3, 79, 18, (1965-1976), (2019).
- Maria-Victoria Mateos, Jesus San-Miguel, Hartmut Goldschmidt, Pieter Sonneveld, Meletios A. Dimopoulos, Bart Heeg, Mahmoud Hashim, William Deraedt, Peter Hu, Annette Lam, Jianming He, The effects of different schedules of bortezomib, melphalan, and prednisone for patients with newly diagnosed multiple myeloma who are transplant ineligible: a matching-adjusted indirect comparison, Leukemia & Lymphoma, 10.1080/10428194.2019.1675881, 61, 3, (680-690), (2019).