Advancements from the EVOLVE study for assessing real-world experience with eteplirsen, golodirsen and casimersen for the treatment of DMD
Publication: Journal of Comparative Effectiveness Research
Abstract
Aim: Eteplirsen, golodirsen and casimersen are phosphorodiamidate morpholino oligomers (PMOs) that have received, based on biomarker data, accelerated approval from the US FDA for the treatment of Duchenne muscular dystrophy (DMD) in patients with pathogenic variants amenable to 51, 53 and 45 exon skipping, respectively. The objectives of this study were to describe patient demographic and baseline functional characteristics, safety and treatment continuation in patients with DMD who were treated with a commercially available PMO in the US from the ongoing phase IV, multicenter, prospective, observational EVOLVE study. Patients & methods: Patients who received or initiated treatment with a PMO at the time of study enrollment as prescribed by treating physicians as part of routine care were included. Approximately 300 patients will be enrolled across the three PMOs. Results: As of this 2023 interim data report, 161 patients were enrolled: 126 were treated with eteplirsen (enrollment complete), mean (standard deviation [SD]) age 14.0 (5.51) years; 23 received golodirsen, mean (SD) age 13.3 (4.25) years; and 12 were treated with casimersen, mean (SD) age 16.1 (7.21) years. Mean (SD) total duration of treatment was 6.2 (1.92) years for eteplirsen, 2.4 (0.83) years for golodirsen and 1.7 (0.62) years for casimersen. All PMOs demonstrated favorable safety profiles, with no treatment-emergent serious adverse events related to treatment. Most patients taking eteplirsen (95.2%, n = 120) continued treatment. Among the 85 patients who were ambulatory at treatment initiation, 37 patients lost ambulation, 34 (91.9%) of whom remained on eteplirsen. Conclusion: Consistent with the safety findings from previous clinical trials, eteplirsen, golodirsen and casimersen showed favorable safety profiles in patients with DMD in routine clinical practice. EVOLVE will continue to describe long-term clinical outcomes.
Clinical Trial Registration Number: NCT06606340
Plain language summary: assessing real-world experience with eteplirsen, golodirsen and casimersen
What is this article about?
Duchenne muscular dystrophy (DMD) is a rare genetic disease in which there is a lack of a muscle protein called dystrophin. Because dystrophin protein is needed for muscle strength and function, over time, people lose the ability to walk and develop breathing and heart problems. In this ongoing real-world study named EVOLVE, investigators collected data from individuals with DMD who were receiving eteplirsen, golodirsen or casimersen from routine clinical visits. EVOLVE is the first and largest study to gather information on the safety and effectiveness of these approved therapies in individuals with DMD. This article includes safety results.
What were the results?
This interim data report included 126 people receiving eteplirsen, 23 receiving golodirsen and 12 receiving casimersen. The results of this study mainly focused on eteplirsen usage, with individuals receiving treatment for 6.2 years at an average age of 14 years. There were no new safety concerns, and most individuals continued using eteplirsen while in the study.
What do the results mean?
The safety results of eteplirsen in this real-world study are similar to those of previous studies of eteplirsen. Collection of data at the start of a study helps investigators understand the types of individuals who receive these medicines in a real-world setting. It is valuable for individuals with DMD to continue completing tests that measure breathing and heart function so that researchers can evaluate how these medicines may change these important functions.
Duchenne muscular dystrophy (DMD) is an X-linked, progressive neuromuscular disease caused by pathogenic variants in the DMD gene that result in the absence of functional dystrophin protein [1,2]. Key disease milestones include loss of ambulation (LOA) [3,4], loss of upper limb function, cardiopulmonary decline with ventilation support [5,6] and early mortality [1,7,8]. Although there is genotype-dependent heterogeneity in disease progression, LOA occurs at approximately 12–14 years in patients receiving corticosteroids [3,4]. With comprehensive medical management of orthopedic, respiratory and cardiac complications, patients can have a life expectancy into their twenties or thirties [1,9,10].
In the US, approved therapies for DMD include phosphorodiamidate morpholino oligomers (PMOs) for patients with exon 51 (eteplirsen), 53 (golodirsen, viltolarsen) or 45 (casimersen) skip-amenable DMD [11–14]; the adeno-associated rh74-viral vector-based gene therapy delandistrogene moxeparvovec [15]; the histone deacetylase inhibitor givinostat [16]; and the glucocorticoids deflazacort and vamorolone [17,18]. Based on an increase in dystrophin production in skeletal muscle, eteplirsen, golodirsen, viltolarsen and casimersen received FDA accelerated approval for the treatment of DMD in September 2016, December 2019, August 2020 and February 2021, respectively. In the US, traditional approvals based on clinical outcomes were granted to deflazacort (2017), vamorolone (2023) and givinostat (2024) [19,20]. Delandistrogene moxeparvovec initially received US approval in 2023 through the accelerated pathway followed by traditional approval in 2024 [21]. Previously published placebo-controlled and open-label clinical trials have demonstrated that PMOs showed dystrophin production and localization [22–24], with a slowed rate of LOA [25–27] and decline in respiratory function [27–30] in patients with DMD compared with genotype-matched natural history controls. Declines in left ventricular ejection fraction (LVEF), indicative of cardiac function decline, were significantly attenuated in eteplirsen-treated individuals compared with matched natural history controls, which suggests a clinical benefit [31]. Survival analyses have indicated a median survival age of 32.8 years in eteplirsen-treated patients compared with 27.4 years in age-matched natural history controls [32]. Studies have also shown that PMOs are well tolerated, with a favorable safety profile [22–25,30,33–35].
EVOLVE (NCT06606340) is a phase IV, multicenter, prospective, observational study to collect retrospective medical history and prospective data on patients with DMD receiving commercial eteplirsen, golodirsen or casimersen in routine clinical practice [36]. The 5-year follow-up period in EVOLVE will collect data regarding exposure to eteplirsen, golodirsen or casimersen, as well as the clinical course of patients with DMD receiving these PMOs. Emerging evidence reported to date from EVOLVE suggests safety profiles of PMOs used commercially are comparable to those in clinical trials [37–39].
The objectives of this interim data report are to provide an in-depth description of EVOLVE study methodology, outline of baseline patient demographic and clinical characteristics, safety and treatment patterns in patients receiving commercially approved PMOs. Describing baseline patient characteristics, including functional data, highlights the heterogeneity of patients who are receiving PMO treatments in the real world.
Patients & methods
This ongoing study is being conducted in accordance with the ethical principles of the Declaration of Helsinki and local regulations for each participating site. The study protocol was reviewed and approved by the appropriate institutional review board and regulatory authority. The patients or patients’ parent(s) or legal guardian(s) provided written informed consent or assent before involvement in any study procedures.
Patients initiated or continued treatment with eteplirsen, golodirsen or casimersen at the time of enrollment as prescribed by treating physicians as part of routine care. Patients enrolling could be either new initiators of PMO therapies (treatment initiation within 6 months of enrollment) or continuing therapy from commercial or prior clinical trial experience. Baseline data refer to either data collected at study entry for those patients continuing therapy or data collected prior to treatment for new PMO initiators.
A total of approximately 300 patients will be enrolled across the three PMOs. The eteplirsen study arm is closed with a total of 126 patients enrolled. Enrollment of DMD patients on golodirsen or casimersen is ongoing. The first of these two treatment groups to reach 100 patients will then be closed to ensure a minimum of 72 patients in the other treatment group. In addition, the protocol requires an enrollment target of 55 new PMO initiators for both the golodirsen and casimersen cohorts to ensure that a minimum number of EVOLVE patients will have baseline data. The study follow-up duration is 5 years for each patient (Figure 1). Enrolled patients who have discontinued PMO treatment will continue to be followed for the duration of this study, in the absence of death or withdrawal of consent.
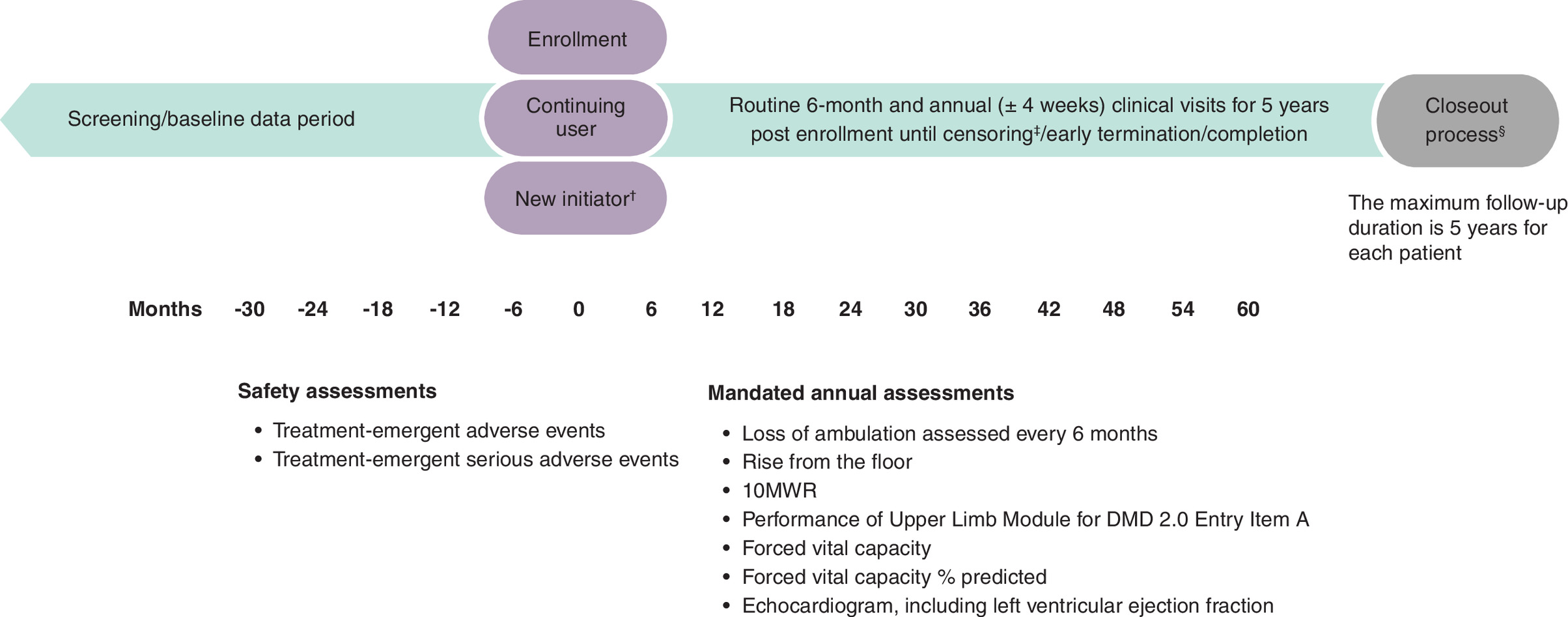
Figure 1. Study schema.
†New initiators must start treatment 6 months before or after enrollment. All patients must start treatment within 6 months of enrollment. The estimated target for golodirsen and casimersen patients to meet ‘New Initiator’ criteria is ≥65%. This will be assessed monthly by data management if 45% of continuing user quota is met.
‡Censoring is earliest of exposure to another DMD gene therapy, new participation in DMD clinical trial, death, disenrollment, loss to follow-up, 1853 days of follow-up (5 years + 28-day visit window) or end of the study period.
§Closeout process triggered by the presence of censoring event or end of follow-up.
10MWR: 10-meter walk/run; DMD: Duchenne muscular dystrophy.
Key inclusion criteria include male at birth, an established clinical diagnosis of DMD (as documented by a genetic report prior to screening), and receiving or initiating treatment with eteplirsen, golodirsen or casimersen at the time of enrollment. Both ambulatory and nonambulatory patients could be enrolled in this study. New initiator patients must have started PMO therapy within 6 months of the date of enrollment. Patients would be excluded if they were participating in any DMD interventional study at the time of enrollment or began participating in another interventional study after enrolling in EVOLVE.
Core outcome data
Seven assessments are mandated in the EVOLVE study (Table 1). However, the treating physician has full discretion for all decisions concerning the patient’s treatment and care. The reasons for not completing assessments are therefore a key part of the dataset. The protocol was amended between 2021 and 2023 to streamline the protocol/electronic data capture (EDC) to reduce patient and healthcare provider burden, and to focus on the core clinical end points.
| Assessment | Description | Frequency of assessment |
|---|---|---|
| Functional outcome assessments | ||
| LOA | • LOA was defined as participant- or caregiver-reported age at continuous wheelchair use, approximated to the nearest month, and verified by an attending physician • LOA was not to be secondary to acute worsening of mobility due to orthopedic morbidity (e.g., fracture, sprain, or other injury) or surgical procedure • LOA may have been determined by an NSAA walk score of 0 (if data on NSAA walk score were available) and/or inability to perform 10MWR | Baseline/enrollment, visits every 6 months, completion/early termination visit |
| Rise from the floor | • Time to rise from the floor is item 11 of the NSAA • The investigator could have stopped the rise from the floor assessment if the patient required >20 s to complete | Baseline/enrollment, annual visits, completion/early termination visit |
| 10MWR | • 10MWR is item 17 of the NSAA • The investigator could have stopped the 10MWR assessment if the patient required >30 s to complete | Baseline/enrollment, annual visits, completion/early termination visit |
| PUL 2.0 Entry Item A | • PUL 2.0 includes an Entry Item A to define the broad starting functional level • Brooke Upper Extremity Scale data were collected in some cases before the EVOLVE protocol was streamlined to only include PUL 2.0 Entry Item A | Baseline/enrollment, annual visits, completion/early termination visit |
| Clinical outcome assessments | ||
| FVC | Pulmonary function testing was conducted using standard spirometry procedures | Baseline/enrollment, annual visits, completion/early termination visit |
| FVC%p | Pulmonary function testing was conducted using standard spirometry procedures | Baseline/enrollment, annual visits, completion/early termination visit |
| Echocardiogram | Echocardiogram included assessment of left ventricular ejection fraction | Baseline/enrollment, annual visits, completion/early termination visit |
10MWR: 10-meter walk/run; DMD: Duchenne muscular dystrophy; FVC: Forced vital capacity; FVC%p: Forced vital capacity % predicted; LOA: Loss of ambulation; NSAA: North Star Ambulatory Assessment; PUL 2.0: Performance of Upper Limb Module for DMD 2.0.
Functional outcomes include LOA, rise from the floor, 10-meter walk/run (10MWR) and Performance of Upper Limb Module for DMD 2.0 (PUL 2.0) Entry Item A (Table 1). A prior protocol version allowed for PUL 2.0 Entry Item A or Brooke Upper Extremity Scale (Brooke) [40]. Rise from the floor, 10MWR and PUL 2.0 Entry Item A were assessed at baseline/enrollment, annual visits and completion/early termination visit. To minimize patient burden during clinical assessments, limits were placed in the EVOLVE protocol to allow the investigator to end the assessment if the patient required >20 s to complete the rise from the floor or >30 s to complete the 10MWR [41,42]. Clinical outcomes include pulmonary function, as measured by forced vital capacity (FVC) and forced vital capacity % predicted (FVC%p), and echocardiogram, which includes assessment of LVEF (Table 1).
Safety outcomes include treatment-emergent adverse events and treatment-emergent serious adverse events (TESAEs) and are assessed at baseline/enrollment, midyear visits (months 6, 18, 30, 42 and 54), annual visits (months 12, 24, 36, 48 and 60) and the completion/early termination visit. A TESAE is defined as an unexpected medical event that at any dose is life threatening, requires inpatient hospitalization or prolongation of existing hospitalization, results in significant or persistent disability/incapacity, results in death, is a congenital anomaly/birth defect, or is a medically important event or reaction. All adverse events, from the signing of informed consent/assent through the last follow-up visit, are recorded in each enrolled participant’s electronic case report form. All adverse events considered related to eteplirsen, golodirsen or casimersen, regardless of seriousness, are also recorded and reported within 24 h of awareness.
Statistical analysis
All results presented are descriptive and not comparative. Descriptive summaries were prepared for continuous variables (mean, median, standard deviation [SD], first quartile, third quartile, minimum/maximum) and categorical variables (number and percentage of nonmissing observations within each category). Demographics and baseline characteristics by treatment were reported using descriptive statistics. Functional and clinical end points by treatment were described using summary statistics. Stratified results for age and ambulatory status are also shown. All tables for this analysis are presented by treatment type.
Results
Patient disposition
The study commenced on 7 January 2019 [36]. As of this interim data report (2 October 2023), 161 patients were enrolled, with 126, 23 and 12 patients receiving eteplirsen, golodirsen and casimersen, respectively (Figure 2) across 21 clinical sites in the United States. The eteplirsen treatment group is fully enrolled. A total of 133 patients were continuing the study and 28 patients had discontinued due to withdrawal by patient (n = 6), deceased (n = 8), protocol violation (n = 3), lost to follow-up (n = 1) and other reasons (n = 10; Figure 2).
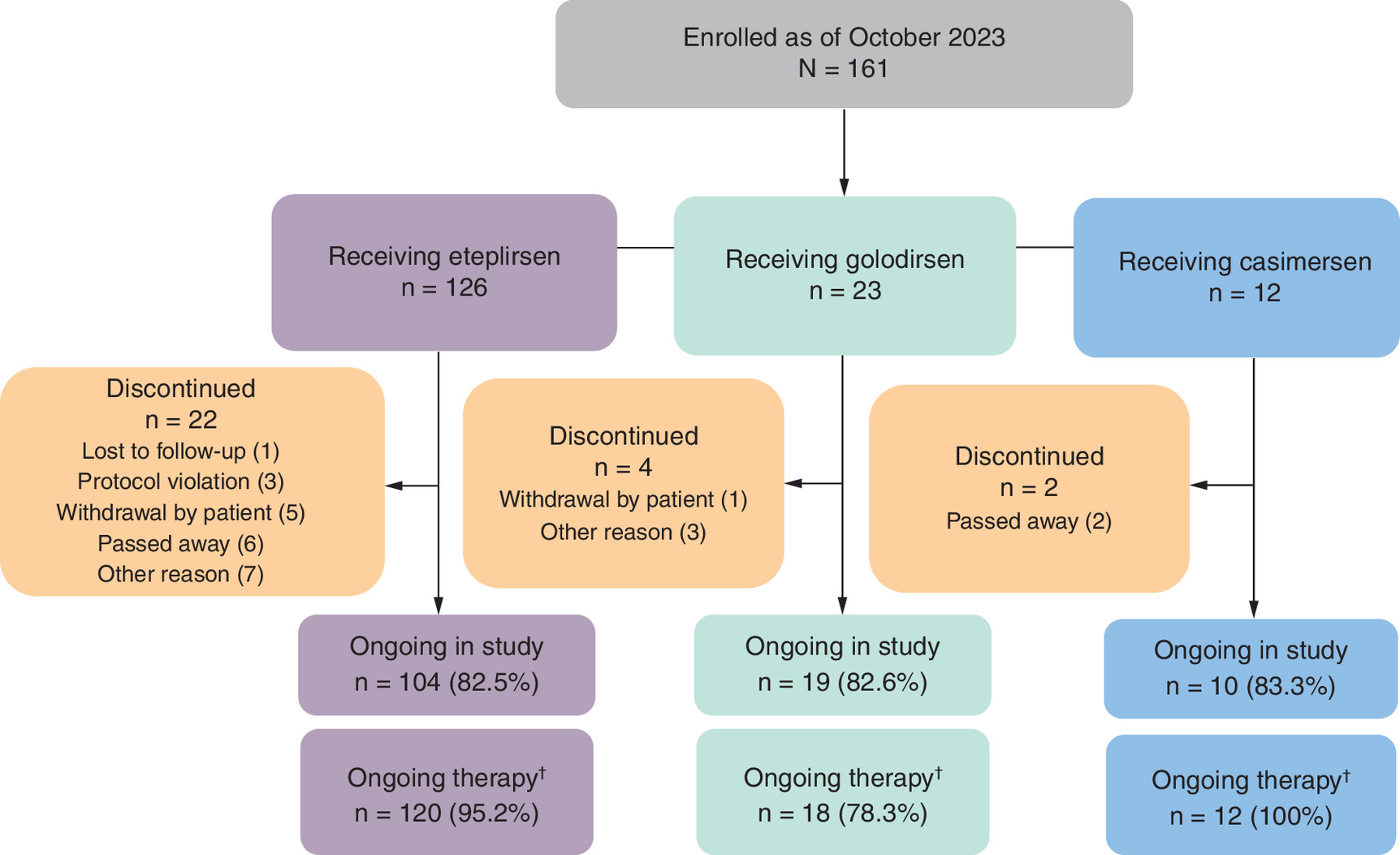
Figure 2. Patient disposition in the EVOLVE study.
†Defined by having not stopped treatment as of last available visit.
Study entry characteristics
Across the three PMOs, patient age at treatment initiation ranged from 1 to 33 years, with a mean age at PMO initiation of 10.7 years for eteplirsen, 12.6 years for golodirsen and 15.8 years for casimersen (Table 2). The mean age at study enrollment was 14.0, 13.3 and 16.1 years for eteplirsen, golodirsen and casimersen, respectively. The mean duration of PMO treatment at study enrollment was 3.4 years for eteplirsen, 0.7 years for golodirsen and 0.3 years for casimersen (Table 2). The mean total duration of PMO treatment was 6.2 years for eteplirsen, 2.4 years for golodirsen and 1.7 years for casimersen (Table 2).
| Parameter | Eteplirsen (n = 126) | Golodirsen (n = 23) | Casimersen (n = 12) |
|---|---|---|---|
| Age at PMO initiation, years Mean (SD) Range | 10.7 (5.13) 1–24 | 12.6 (4.31) 4–19 | 15.8 (7.23) 6–33 |
| Age at study enrollment, years Mean (SD) Range | 14.0 (5.51) 1–28 | 13.3 (4.25) 6–20 | 16.1 (7.21) 6–33 |
| Race, n (%) White Other/missing | 96 (76.2) 30 (23.8) | 18 (78.3) 5 (21.7) | 10 (83.3) 2 (16.7) |
| Ethnicity, n (%) Hispanic or Latino Not Hispanic or Latino Not reported/unknown | 27 (21.4) 86 (68.3) 13 (10.3) | 6 (26.1) 14 (60.9) 3 (13.0) | 2 (16.7) 10 (83.3) 0 |
| BMI, kg/m2, mean (SD) | 24.1 (8.2)† | 22.3 (5.3)‡ | 25.7 (6.6)§ |
| Nonambulatory, n (%) At PMO initiation At enrollment At last available visit | 40 (31.7) 71 (56.3) 77 (61.1) | 12 (52.2) 15 (65.2) 16 (69.6) | 9 (75.0) 9 (75.0) 9 (75.0) |
| PMO use, n (%) New initiator¶ Prevalent# | 8 (6.3) 118 (93.7) | 11 (47.8) 12 (52.2) | 9 (75.0) 3 (25.0) |
| PMO treatment, mean (SD), years Total duration†† At study enrollment‡‡ | 6.2 (1.92) 3.4 (1.90) | 2.4 (0.83) 0.7 (0.68) | 1.7 (0.62) 0.3 (0.31) |
| Treatment status as of last available visit, n (%) Continuing therapy§§ Stopped (discontinued) | 120 (95.2) 6 (4.8) | 18 (78.3) 5 (21.7) | 12 (100) 0 |
| PMO treatment in previous clinical trial, n (%) | 40 (31.7) | 1 (4.3) | 0 |
| Corticosteroid use, n (%) Prior to PMO initiation At or after PMO initiation In the past 12 months prior to study enrollment | 76 (60.3) 114 (90.5) 112 (88.9) | 20 (87.0) 21 (91.3) 21 (91.3) | 10 (83.3) 11 (91.7) 10 (83.3) |
| Age at documented first use of corticosteroid treatment, years n Mean (SD) Range | 116 8.9 (4.3) 1.7–22.9 | 21 8.2 (3.6) 3.0–16.4 | 11 10.6 (7.2) 4.7–29.7 |
†
n = 92.
‡
n = 17.
§
n = 7.
¶
New initiators started eteplirsen, golodirsen or casimersen treatment within 6 months of enrolling in EVOLVE.
#
Prevalent users initiated eteplirsen, golodirsen or casimersen treatment more than 6 months prior to enrollment in EVOLVE.
††
Total duration of eteplirsen/golodirsen/casimersen treatment (years) = ([treatment end date - treatment start date] + 1)/365.25. If treatment end date was missing, then cutoff date was used.
‡‡
Total duration of eteplirsen/golodirsen/casimersen treatment at study enrollment (years) = ([study enrollment date - treatment start date] + 1)/365.25.
§§
Defined by having not stopped treatment as of last available visit.
BMI: Body mass index; PMO: Phosphorodiamidate morpholino oligomer; SD: Standard deviation.
At PMO initiation, 31.7% (n = 40), 52.2% (n = 12), and 75.0% (n = 9) of patients receiving eteplirsen, golodirsen and casimersen, respectively, were nonambulatory (Table 2). The percentage of patients that were nonambulatory at study enrollment was 56.3% (n = 71) for eteplirsen, 65.2% (n = 15) for golodirsen and 75.0% (n = 9) for casimersen (Table 2).
Most patients (96.9% [n = 156]) reported receiving concomitant medications during the EVOLVE study (Supplementary Table 1). The most common concomitant medications (excluding corticosteroids) were vitamin D and analogues (78.9% [n = 127]), angiotensin-converting enzyme (ACE) inhibitors (73.3% [n = 118]), aldosterone antagonists (46.0% [n = 74]), multivitamins (26.7% [n = 43]) and osmotically active laxatives (25.5% [n = 41]).
Almost all patients receiving eteplirsen (98.4% [n = 124]) and golodirsen (95.7% [n = 22]) and all patients receiving casimersen (100% [n = 12]) had comorbidities reported at or before EVOLVE study entry (Supplementary Table 2). The most common comorbidities reported in patients receiving eteplirsen were vitamin D deficiency (26.2% [n = 33]), constipation (23.0% [n = 29]) and restrictive lung disease (19.8% [n = 25]). Vitamin D deficiency (21.7% [n = 5]), attention-deficit hyperactivity disorder (17.4% [n = 4]), delayed puberty (17.4% [n = 4]), low bone density (17.4% [n = 4]), obstructive sleep apnea (17.4% [n = 4]) and speech delay (17.4% [n = 4]) were the most frequent comorbidities in patients receiving golodirsen (Supplementary Table 2). Autism spectrum disorder (25.0% [n = 3]), constipation (25.0% [n = 3]), low bone mineral density (25.0% [n = 3]) and vitamin D deficiency (25.0% [n = 3]) were the most common comorbidities in patients receiving casimersen (Supplementary Table 2). Supplementary Table 2 directly summarizes the medical history as reported by the study site at EVOLVE study entry.
As of this interim data report, 35 patients aged <84 months (<7 years) were enrolled at the time of PMO initiation, with 32 patients, two patients and one patient receiving eteplirsen, golodirsen and casimersen, respectively (Figure 3). Most patients receiving eteplirsen (81.3% [n = 26]) and all patients receiving golodirsen (n = 2) and casimersen (n = 1) remain in the study.
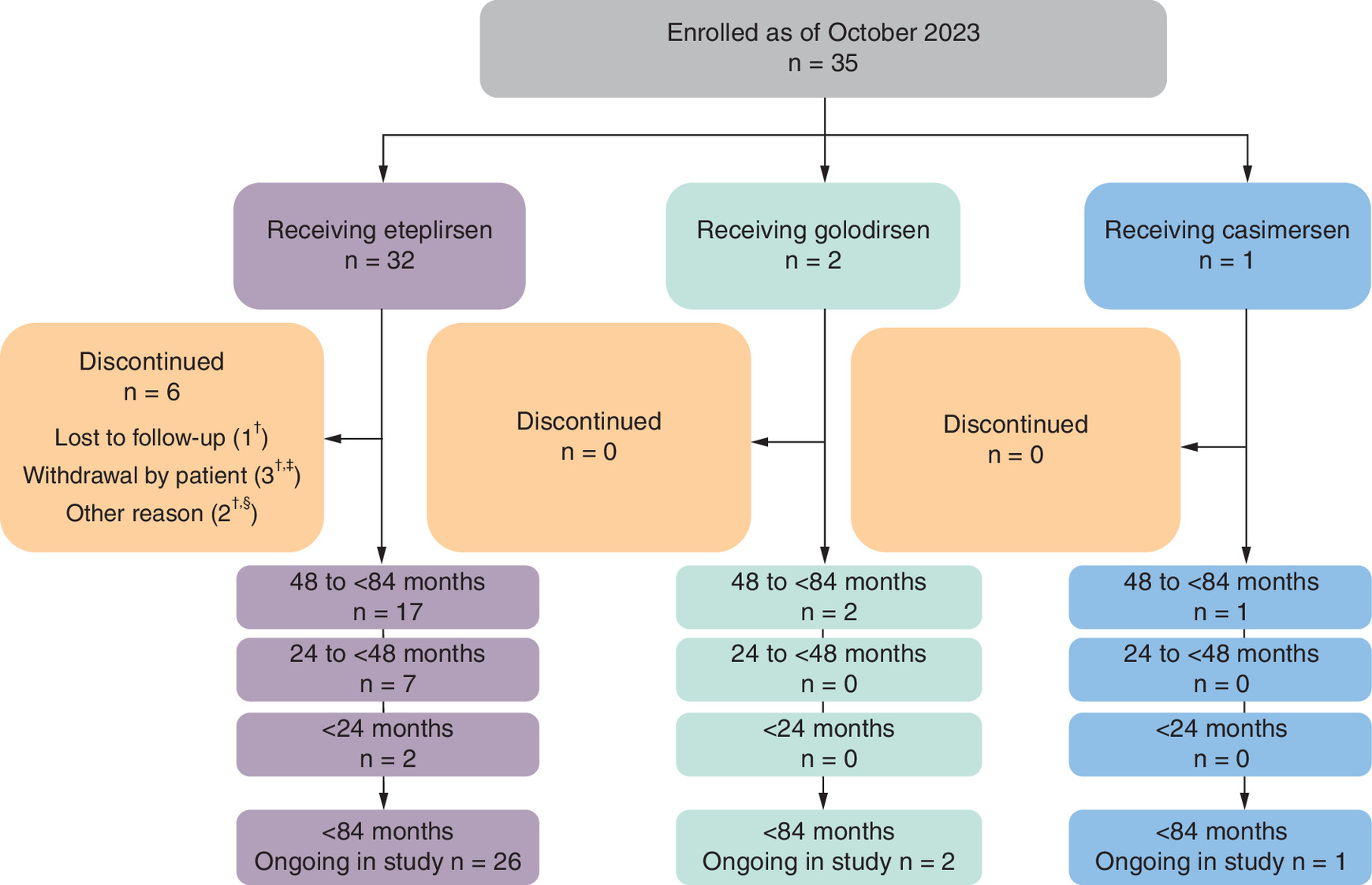
Figure 3. Young patient disposition in the EVOLVE study (age at PMO treatment initiation <84 months).
†Four patients in the 48- to <84-month group receiving eteplirsen discontinued due to patient withdrawal (n = 2), were lost to follow-up (n = 1) or for other reasons (n = 1).
‡One patient in the <24-month group receiving eteplirsen discontinued due to patient withdrawal.
§One patient in the 24- to <48-month group receiving eteplirsen discontinued due to other reasons.
PMO: Phosphorodiamidate morpholino oligomer.
In patients aged between 48 and <84 months receiving eteplirsen (n = 21), mean (SD) age at PMO initiation was 5.8 (0.77) years, age at study enrollment was 8.9 (1.54) years and time from DMD diagnosis to PMO initiation was 2.2 (1.76) years (Table 3). Total duration of PMO treatment was 5.9 (1.79) years and duration of PMO treatment at study enrollment was 3.1 (1.33) years (Table 3). Among patients receiving eteplirsen, all patients aged <48 months (n = 11) were ambulatory. In patients aged between 48 and <84 months, 95.2% (n = 20) were ambulatory and 4.8% (n = 1) were nonambulatory at PMO initiation. One patient aged 48 to <84 months who was nonambulatory at treatment initiation used corticosteroids after PMO treatment initiation (Table 3). For patients receiving golodirsen (n = 2) aged between 48 and <84 months, mean (SD) age at PMO initiation was 5.6 (1.23) years and age at study enrollment was 6.9 (0.30) years. The casimersen-treated patient (n = 1) initiated PMO therapy at 6.2 years of age and was enrolled in the study at 6.7 years of age.
| Parameter | Aged <24 months (n = 3) | Aged 24 to <48 months (n = 8) | Aged 48 to <84 months (n = 24) | ||
|---|---|---|---|---|---|
| Eteplirsen (n = 3) | Eteplirsen (n = 8) | Eteplirsen (n = 21) | Golodirsen (n = 2) | Casimersen (n = 1) | |
| Age at PMO initiation, mean (SD), years | 1.78 (0.05) | 3.34 (0.44) | 5.77 (0.77) | 5.56 (1.23) | 6.24 |
| Age at study enrollment, mean (SD), years | 3.47 (1.56) | 4.82 (1.05) | 8.86 (1.54) | 6.85 (0.30) | 6.68 |
| Time from DMD diagnosis to PMO initiation, mean (SD), years | 0.28 (0.07) | 1.04 (1.20) | 2.23 (1.76) | 3.13 (1.74) | 1.11 |
| Ambulatory status at baseline, n (%) Ambulatory Nonambulatory | 3 (100.0) 0 | 8 (100.0) 0 | 17 (81.0) 4 (19.0) | 2 (100.0) 0 | 1 (100.0) 0 |
| PMO treatment, mean (SD), years Total duration At study enrollment | 4.30 (1.53) 1.70 (1.51) | 4.21 (1.85) 1.48 (1.01) | 5.91 (1.79) 3.10 (1.33) | 1.34 (0.94) 1.29 (0.93) | 2.47 0.45 |
| Corticosteroid use, n (%) Prior to PMO initiation At or after PMO initiation | 0 1 (33.3) | 2 (25.0) 6 (75.0) | 11 (52.4) 20 (95.2) | 1 (50.0) 2 (100.0) | 0 1 (100.0) |
DMD: Duchenne muscular dystrophy; PMO: Phosphorodiamidate morpholino oligomer; SD: Standard deviation.
Treatment continuation
Patients remaining on treatment was defined by patients having not stopped treatment as of the last available study visit. Patients continuing on eteplirsen in the EVOLVE study was 95.2% (n = 120) over an average of 6.2 years from treatment initiation (Table 2 & Figure 2). A small percentage of patients (4.8% [n = 6]) discontinued eteplirsen for reasons not due to adverse events. Similarly, 78.3% (n = 18) of patients remained on golodirsen and 100% (n = 12) of patients remained on casimersen as of the interim data cutoff (Table 2 & Figure 2).
Corticosteroid use
Most patients (88.8% [n = 143]) had received corticosteroid treatment in the previous 12 months (Supplementary Table 3). Prior to enrollment, the mean age of corticosteroid initiation was 8.9 years, with the youngest patient aged 1.7 years and oldest patient aged 29.7 years. The most common corticosteroids at any time prior to enrollment were prednisone among eteplirsen-treated patients (46.0% [n = 58]) and deflazacort among golodirsen-treated patients (47.8% [n = 11]) and casimersen-treated patients (58.3% [n = 7]).
Based on follow-up data capturing new or a change in corticosteroid treatment after enrollment, 28.6% (n = 36) of eteplirsen-treated patients, 17.4% (n = 4) of golodirsen-treated patients and 25.0% (n = 3) of casimersen-treated patients reported new or change in corticosteroid treatment after enrollment (Supplementary Table 3). Mean age at first corticosteroid capture after enrollment for all patients was 12.2 years, with the youngest patient aged 4.7 years and the oldest patient aged 21.7 years. The most common corticosteroid was deflazacort among eteplirsen-treated patients (19.0% [n = 24]) and prednisone among golodirsen-treated patients (17.4% [n = 4]) and casimersen-treated patients (25.0% [n = 3]). Five (4.0%) eteplirsen-treated patients and one (8.3%) casimersen-treated patient switched from prednisone to deflazacort, and three (2.4%) eteplirsen-treated patients switched from deflazacort to prednisone.
No patient aged <24 months at initiation (n = 3) had corticosteroid exposure prior to PMO treatment. Of eight patients initiating eteplirsen aged between 24 and <48 months, two patients received corticosteroid (prednisone) before eteplirsen initiation. Six patients commenced corticosteroid at or after eteplirsen initiation, with a mean age at corticosteroid initiation of 51 months (4.25 years) (Supplementary Table 4). Of 24 patients initiating PMO aged between 48 and <84 months, 12 patients commenced corticosteroid (prednisone n = 9, deflazacort n = 3) before PMO initiation (eteplirsen n = 11, golodirsen n = 1) and 23 patients received corticosteroid at or after PMO initiation, with a mean age at corticosteroid initiation of 73 months (6.1 years) (Supplementary Table 4).
Safety
PMOs demonstrated favorable safety profiles and were well tolerated as no TESAEs were found related to eteplirsen, golodirsen or casimersen treatment. Exposure-adjusted incidence rate per 100 subject-years (standard error [SE]) remained flat over time for eteplirsen 11.8 (1.99), golodirsen 5.5 (3.91) and casimersen 36.5 (16.34).
Eteplirsen was well tolerated in young patients initiating treatment at <84 months (<7 years). TESAEs in younger patients included acute myocarditis, catheter-site erythema, pyrexia, fracture and sickle cell crisis (reported twice in the same patient). None of the TESAEs were determined to be related to treatment.
Port use in young patients was reported in the physician notes or adverse event listing in at least 23 of 32 (71.9%) eteplirsen-treated patients, one of two (50%) golodirsen-treated patients and one (100%) casimersen-treated patient; patients as young as 20 months received a port. As port use was not a mandatory collected observation, this may be an underestimation of port use in this population. Six patients experienced a total of nine port-related adverse events. One port-related adverse event was considered related to PMO treatment (device positioned inappropriately); one was serious, and seven were mild in severity. There were no port-related infections related to treatment.
Eteplirsen-specific safety
In the fully enrolled eteplirsen cohort (n = 126), the most frequent TESAEs by system organ class were infections and infestations (n = 12), injury, poisoning and procedural complications (n = 12), cardiac disorders (n = 6) and respiratory, thoracic and mediastinal disorders (n = 6; Table 4). The most common TESAEs related to infection and infestations were pneumonia (n = 5), COVID-19 (n = 2) and septic shock (n = 2); related to injury, poisoning and procedural complications was femur fracture (n = 7); and related to respiratory, thoracic and mediastinal disorders was acute respiratory failure (n = 4).
| Eteplirsen (n = 126) | Golodirsen (n = 23) | Casimersen (n = 12) | |
|---|---|---|---|
| Any TESAE, n/N (%) | 35/126 (27.8) | 2/23 (8.7) | 5/12 (41.7) |
| Year 1 | 14/126 (11.1) | 0/23 (0) | 3/12 (25.0) |
| Year 2 | 12/122 (9.8) | 1/19 (5.3) | 2/11 (18.2) |
| Year 3 | 12/115 (10.4) | 1/11 (9.1) | 0/1 (0) |
| Year 4 | 7/64 (10.9) | 0/3 (0) | – |
| Year 5 | 0/16 (0) | – | – |
| TESAEs by system organ class, n (%) | |||
| Blood/lymphatic disorders Iron deficiency anemia Sickle cell anemia with crisis | 2 (1.6) 1 (0.8) 1 (0.8) | 0 0 0 | 0 0 0 |
| Cardiac disorders Acute myocardial infarction Atrial flutter Cardiac arrest Cardiac disorder Cardiac failure acute Cardiorespiratory arrest Myocarditis Pericardial effusion Pulseless electrical activity | 6 (4.8) 1 (0.8) 1 (0.8) 0 1 (0.8) 1 (0.8) 1 (0.8) 1 (0.8) 0 1 (0.8) | 0 0 0 0 0 0 0 0 0 0 | 2 (16.7) 0 0 1 (8.3) 0 0 0 0 1 (8.3) 0 |
| Gastrointestinal disorders Dysphagia Eosinophilic esophagitis Gastritis Impaired gastric emptying Intestinal obstruction Large intestine perforation Tooth impacted Volvulus | 4 (3.2) 1 (0.8) 0 0 1 (0.8) 1 (0.8) 1 (0.8) 1 (0.8) 1 (0.8) | 1 (4.3) 0 1 (4.3) 0 0 0 0 0 0 | 1 (8.3) 0 0 1 (8.3) 0 0 0 0 0 |
| General disorders and administration site conditions Catheter-site erythema Pyrexia | 2 (1.6) 1 (0.8) 2 (1.6) | 0 0 0 | 0 0 0 |
| Infections and infestations COVID-19 Campylobacter gastroenteritis Device-related infection Device-related sepsis Pneumonia Pneumonia aspiration Pseudomonal bacteremia Pulmonary sepsis Sepsis Septic shock | 12 (9.5) 2 (1.6) 1 (0.8) 1 (0.8) 1 (0.8) 5 (4.0) 1 (0.8) 1 (0.8) 1 (0.8) 1 (0.8) 2 (1.6) | 1 (4.3) 1 (4.3) 0 0 0 0 0 0 0 0 0 | 2 (16.7) 0 0 0 0 0 0 0 0 2 (16.7) 0 |
| Injury, poisoning and procedural complications Craniocerebral injury Femur fracture Fibula fracture Gastrostomy tube site complication Humerus fracture Lower limb fracture Lumbar vertebral fracture Tibia fracture Vascular pseudoaneurysm Wound dehiscence | 12 (9.5) 1 (0.8) 7 (5.6) 1 (0.8) 1 (0.8) 1 (0.8) 1 (0.8) 1 (0.8) 1 (0.8) 1 (0.8) 1 (0.8) | 0 0 0 0 0 0 0 0 0 0 0 | 0 0 0 0 0 0 0 0 0 0 0 |
| Investigations Troponin increased Weight decreased | 2 (1.6) 1 (0.8) 1 (0.8) | 0 0 0 | 0 0 0 |
| Metabolism and nutrition disorders Hypokalemia | 1 (0.8) 1 (0.8) | 0 0 | 0 0 |
| Musculoskeletal and connective tissue disorders Neuromuscular scoliosis Osteoporosis Scoliosis Tendinous contracture | 4 (3.2) 1 (0.8) 1 (0.8) 1 (0.8) 1 (0.8) | 0 0 0 0 0 | 0 0 0 0 0 |
| Product issues† Device issue Device malfunction | 2 (1.6) 1 (0.8) 1 (0.8) | 0 0 0 | 0 0 0 |
| Psychiatric disorders Hallucination Psychotic disorder Suicide attempt | 2 (1.6) 1 (0.8) 1 (0.8) 1 (0.8) | 0 0 0 0 | 0 0 0 0 |
| Renal and urinary disorders | 0 | 0 | 1 (8.3) |
| Respiratory, thoracic and mediastinal disorders Acute respiratory failure Dyspnea Pleural effusion Pneumonitis aspiration Pulmonary embolism Respiratory distress | 6 (4.8) 4 (3.2) 1 (0.8) 1 (0.8) 1 (0.8) 1 (0.8) 1 (0.8) | 0 0 0 0 0 0 0 | 1 (8.3) 0 0 1 (8.3) 0 0 0 |
| Vascular disorders Hypotension Shock | 2 (1.6) 2 (1.6) 0 | 0 0 0 | 1 (8.3) 0 1 (8.3) |
| Ambulatory at enrollment, EAIR/100 years (SE) Any TESAE | 7.8 (2.4) | 8.6 (8.6) | 20.7 (20.7) |
| Nonambulatory at enrollment, EAIR/100 years (SE) Any TESAE | 15.4 (3.1) | 4.1 (4.1) | 45.2 (22.6) |
†
Port accessibility.
EAIR/100 years: Exposure-adjusted incidence rate per 100 subject-years; PMO: Phosphorodiamidate morpholino oligomer; SE: standard error; TESAE: Treatment-emergent serious adverse event.
Among the eteplirsen-treated patients who were ambulatory at enrollment, three patients had a femur fracture (5.5%), one (1.8%) patient had a lower limb fracture and one (1.8%) patient had a lumbar vertebral fracture. These events were considered not related to treatment.
Of the eteplirsen-treated patients who were nonambulatory at enrollment, one (1.4%) patient had a fibular fracture (year 3), one (1.4%) had a humeral fracture (year 3), one (1.4%) had a tibial fracture (year 1), and four (two [2.8%] in year 1; two [3.2%] in year 3) had femoral fractures. All TESAEs were determined to be not related to treatment.
Study entry characteristics by ambulatory status in eteplirsen-treated patients
Among 126 eteplirsen-treated patients, 48 (38.1%) were ambulatory at eteplirsen initiation and through follow-up, 41 (32.5%) were nonambulatory at treatment initiation, and 37 (29.4%) lost ambulation after eteplirsen initiation (85 patients were ambulatory at treatment initiation; Table 5). Of the 48 (38.1%) patients who were ambulatory at eteplirsen initiation and through follow-up, the mean age was 7.0 years at eteplirsen initiation and 9.8 years at study enrollment (Table 5). The mean duration of eteplirsen treatment at study enrollment was 2.8 years, and the mean total duration of eteplirsen treatment was 5.6 years. Over half (54.2% [n = 26]) of these patients had received corticosteroids prior to eteplirsen initiation, with a mean age of 7.0 years at first use of corticosteroid. Most patients reported receiving corticosteroid treatment in the past 12 months prior to study enrollment (89.6% [n = 43]) and at or after eteplirsen initiation (89.6% [n = 43]; Table 5).
| Parameter | Ambulatory at eteplirsen initiation through follow-up (n = 48) | Nonambulatory at eteplirsen initiation (n = 41) | Nonambulatory after eteplirsen initiation (n = 37) |
|---|---|---|---|
| Age at eteplirsen initiation, years Mean (SD) Range | 7.0 (3.8) 1–20 | 15.4 (4.1) 7–24 | 10.2 (3.3) 5–19 |
| Age at study enrollment, years Mean (SD) Range | 9.83 (4.7) 1–24 | 18.2 (4.1) 10–28 | 14.8 (3.6) 7–23 |
| Age at confirmed diagnosis, years n Mean (SD) Range | 47 3.8 (2.5) 0–11 | 41 5.7 (2.9) 1–15 | 37 4.9 (2.4) 0–9 |
| Time from confirmed diagnosis of DMD to eteplirsen initiation, years n Mean (SD) Range | 47 3.2 (3.4) 0–16 | 41 9.7 (4.2) 2–18 | 37 5.2 (4.1) 0–19 |
| Duration of eteplirsen treatment, mean (SD), years Total duration At study enrollment | 5.6 (2.1) 2.8 (1.8) | 5.7 (1.3) 2.9 (1.3) | 7.4 (1.7) 4.6 (2.0) |
| Treatment status as of last available visit, n (%) Continuing therapy (persistence)† Stopped (discontinued) | 46 (95.8) 2 (4.2) | 40 (97.6) 1 (2.4) | 34 (91.9) 3 (8.1) |
| Corticosteroid use, n (%) Prior to eteplirsen initiation At or after eteplirsen initiation In the past 12 months prior to study enrollment | 26 (54.2) 43 (89.6) 43 (89.6) | 33 (80.5) 34 (82.9) 33 (80.5) | 17 (45.9) 37 (100) 36 (97.3) |
| Age at first use of corticosteroid treatment, years n Mean (SD) Range | 43 7.0 (3.7) 2–21 | 36 10.5 (4.8) 2–22 | 37 9.5 (3.6) 1–19 |
†
Defined as not having stopped treatment as of last available visit.
DMD: Duchenne muscular dystrophy; SD: standard deviation.
Of the 41 (32.5%) patients who were nonambulatory at eteplirsen initiation, mean age was 15.4 years at eteplirsen initiation and 18.2 years at study enrollment (Table 5). The mean duration of eteplirsen treatment at study enrollment was 2.9 years, and the mean total duration of eteplirsen treatment was 5.7 years. Most (80.5% [n = 33]) patients had received corticosteroids prior to eteplirsen initiation, with a mean age of 10.5 years at first use of corticosteroid. Similarly, many patients reported receiving corticosteroid treatment in the past 12 months prior to study enrollment (80.5% [n = 33]) and at or after eteplirsen initiation (82.9% [n = 34]) (Table 5).
Of the 37 (29.4%) patients who were nonambulatory after eteplirsen initiation, the mean age was 10.2 years at eteplirsen initiation and 14.8 years at study enrollment (Table 5). The mean duration of eteplirsen treatment at study enrollment was 4.6 years, and the mean total duration of eteplirsen treatment was 7.4 years. Less than half (45.9% [n = 17]) of patients had received corticosteroids prior to eteplirsen initiation, with a mean age of 9.5 years at first use of corticosteroid. Most patients reported receiving corticosteroid treatment in the past 12 months prior to study enrollment (97.3% [n = 36]) and all patients at or after eteplirsen initiation (100.0% [n = 37]) (Table 5).
Across the subgroups, 95.8% (n = 46) of patients who were ambulatory at treatment initiation remained on eteplirsen, 97.6% (n = 40) who were nonambulatory at treatment initiation continued eteplirsen, and 91.9% (n = 34) of patients who lost ambulation after eteplirsen initiation remained on eteplirsen (Table 5).
In young patients who commenced eteplirsen treatment at 48 to <84 months of age, mean age at treatment initiation was 5.8 years overall (n = 21), 5.5 years in patients who were ambulatory at data cutoff (n = 14), and 6.3 years in patients who were nonambulatory at data cutoff (n = 7; Table 6).
| Parameter | Nonambulatory (n = 7) | Ambulatory (n = 14) | Total (n = 21) |
|---|---|---|---|
| Age at treatment initiation, mean (SD), years | 6.27 (0.63) | 5.51 (0.73) | 5.77 (0.77) |
| Age at first use of corticosteroid treatment, mean (SD), years | 7.72 (1.60) | 5.26 (0.93)† | 6.12 (1.67)‡ |
| Duration of eteplirsen until data cutoff, mean (SD), years | 6.18 (1.48) | 5.77 (1.96) | 5.91 (1.79) |
| Age at data cutoff, mean (SD), years | 12.95 (1.47) | 11.41 (2.10) | 11.92 (2.01) |
†
n = 13.
‡
n = 20.
SD: Standard deviation.
Functional & clinical data for eteplirsen-treated patients at study entry
Mandated study end points were assessed for completeness and study entry descriptive statistics to contextualize the EVOLVE patient population. Assessment data for rise from floor and 10MWR were available for 15.9% (n = 20/126) and 25.4% (n = 32/126), respectively, of patients receiving eteplirsen at study entry. Among all eteplirsen-treated patients, mean (SD) values were 10.63 (13.46) s for rise from floor (n = 20) and 7.09 (3.46) s for 10MWR (n = 32) (Table 7). A rise from floor time >20 s was reported in 4.8% (n = 6) of patients.
| Outcome | Nonambulatory at EVOLVE study entry (n = 71) | Ambulatory at EVOLVE study entry (n = 55) | Total patients at EVOLVE study entry (n = 126) |
|---|---|---|---|
| Rise from the floor, seconds | |||
| n (%) Age, mean (SD), years | 0 | 20 (36.4) 9.03 (2.09) | 20 (15.9) 9.03 (2.09) |
| Mean (SD) Median Min, max† | 0 | 10.63 (13.46) 4.75 2.3, 53.8 | 10.63 (13.46) 4.75 2.3, 53.8 |
| Patients with ≥2 data points, n (%) | – | – | 22 (17.5) |
| 10MWR, seconds | |||
| n (%) Age, mean (SD), years | 1 (1.4) 10.16 (NA) | 31 (56.4) 9.96 (3.95) | 32 (25.4) 9.97 (3.89) |
| Mean (SD) Median Min, max | 9.00 (NA) 9.00 9.0, 9.0 | 7.03 (3.50) 5.70 3.3, 17.0 | 7.09 (3.46) 5.75 3.3, 17.0 |
| Patients with ≥2 data points, n (%) | – | – | 32 (25.4) |
| PUL 2.0 Entry Item A | |||
| n (%) Age, mean (SD), years | 17 (23.9) 15.99 (3.97) | 2 (3.6) 12.48 (3.54) | 19 (15.1) 15.62 (3.99) |
| Mean (SD) Median Min, max | 3.4 (1.90) 4.0 1.0, 6.0 | 3.0 (2.83) 3.0 1.0, 5.0 | 3.3 (1.92) 4.0 1.0, 6.0 |
| Brooke Upper Extremity Scale | |||
| n (%) Age, mean (SD), years | 40 (56.3) 16.34 (4.07) | 24 (43.6) 11.28 (5.32) | 64 (50.8) 14.44 (5.16) |
| Mean (SD) Median Min, max | 3.6 (1.48) 4.0 1.0, 6.0 | 1.6 (0.65) 2.0 1.0, 3.0 | 2.9 (1.57) 2.0 1.0, 6.0 |
| Patients with ≥2 data points, n (%)‡ | – | – | 84 (66.7) |
| FVC%p | |||
| n (%) Age, mean (SD), years | 50 (70.4) 16.78 (3.77) | 31 (56.4) 11.65 (4.07) | 81 (64.3) 14.81 (4.61) |
| Mean (SD) Median Min, max | 52.62 (22.74) 52.13 12.87, 107.80 | 93.21 (20.93) 97.27 47.91, 143.64 | 68.15 (29.58) 67.79 12.87, 143.64 |
| Patients with ≥2 data points, n (%) | – | – | 78 (61.9) |
| LVEF | |||
| n (%) Age, mean (SD), years | 38 (53.5) 17.27 (4.10) | 30 (54.5) 11.16 (5.06) | 68 (54.0) 14.58 (5.45) |
| Mean (SD) Median Min, max | 58.39 (7.71) 58.0 40.0, 76.6 | 60.52 (8.76) 61.50 30.0, 72.0 | 59.33 (8.20) 60.0 30.0, 76.6 |
| Patients with ≥2 data points, n (%) | – | – | 73 (57.9) |
| LVEF <55% | |||
| n (%) Age, mean (SD), years | 8 (11.3) 20.16 (5.84) | 5 (9.1) 13.97 (6.12) | 13 (10.3) 17.78 (6.50) |
| Mean (SD) Median Min, max | 47.40 (5.00) 48.3 40.0, 54.6 | 45.40 (9.63) 46.0 30.0, 54.0 | 46.63 (6.82) 48.0 30.0, 54.6 |
| ECG assessment of LVEF | |||
| n (%) Age, mean (SD), years | – | – | 68 (54.0) 14.58 (5.45) |
| Mean (SD) Median Min, max | – | – | 59.33 (8.20) 60.0 30.0, 76.6 |
†
Investigator could have stopped the assessment if the patient required >20 s to complete.
‡
Combined PUL 2.0 and Brooke Upper Extremity Scale.
10MWR: 10-meter walk/run; DMD: Duchenne muscular dystrophy; ECG: Electrocardiogram; FVC%p: Forced vital capacity % predicted; LVEF: Left ventricular ejection fraction; NA: Not available; PUL 2.0: Performance of Upper Limb Module for DMD 2.0; SD: Standard deviation.
Data for the eteplirsen-treated ambulatory patients at study entry (n = 55) were most complete for rise from floor (36.4%; n = 20/55) and 10MWR (56.4%; n = 31/55). The mean (SD) value for rise from floor for eteplirsen-treated ambulatory patients (n = 20) was 10.63 (13.46) s. The mean (SD) value for 10MWR was 7.03 (3.50) s in 31 eteplirsen-treated ambulatory patients (Table 7).
The combined data availability for both PUL 2.0 Entry Item A and Brooke were available in 66.7% (84/126) of patients receiving eteplirsen at study entry. Median (min, max) PUL 2.0 Entry Item A values were 4.0 (1.0, 6.0) for all patients at study entry (n = 19), 3.0 (1.0, 5.0) in ambulatory patients (n = 2), and 4.0 (1.0, 6.0) in nonambulatory patients (n = 17) (Table 7). Median (min, max) Brooke upper extremity values were 2.0 (1.0, 6.0) for all patients at study entry (n = 64), 2.0 (1.0, 3.0) for ambulatory patients (n = 24), and 4.0 (1.0, 6.0) for nonambulatory patients (n = 40). Mean (SD) FVC%p value for all patients at study entry (n = 81) was 68.15 (29.58). FVC%p values appeared to be higher for ambulatory (93.21 [20.93]; n = 31) versus nonambulatory (52.62 [22.74]; n = 50) patients at study entry (Table 7). Data completeness for LVEF was available in 54.0% (68/126) of patients receiving eteplirsen at EVOLVE study entry. Mean (SD) values for LVEF were 59.33 (8.20) for all patients at study entry (n = 68). Mean (SD) values for LVEF (<55% at study entry) were 46.63 (6.82) for all patients at study entry (n = 13). LVEF (mean [SD]) data were similar for eteplirsen-treated ambulatory patients (60.52 [8.76]; n = 30) and nonambulatory patients (58.39 [7.71]; n = 38). Similar results were observed in LVEF <55% for eteplirsen-treated ambulatory patients (45.40 [9.63]; n = 5) and nonambulatory patients (47.40 [5.00]; n = 8). Among all eteplirsen-treated patients (n = 126), an ECG assessment of LVEF was available in 54.0% (n = 68) of patients at study entry, with a mean age of 14.6 years (Table 7).
Data completeness over EVOLVE inception
Protocol amendments between 2022 and 2023 aimed to streamline the assessments to reduce patient and healthcare provider burden. Completeness of three of the protocol mandated end points was evaluated using a later data cut, September 2024, which were the most comprehensive data available. Upper limb (Brooke score/PUL 2.0 Entry Item A), cardiac (aged ≥6 and ≥12 years) and pulmonary (aged ≥6 and ≥12 years) assessments by calendar year and by subgroups before and after the protocol amendments were evaluated. The number of assessments in each visit was captured and divided by the total number of expected assessments from all patients in the specific year.
Capture of data for upper limb end points across all patients receiving PMOs improved over time from 2021 to 2023, with 20% (n = 50/253) in 2021, 24% (n = 72/297) in 2022 and 37% (n = 100/273). Cardiac and pulmonary end points did not show an increase in completed data over time. For 10MWR and rise from the floor functional outcomes, respectively, at least two repeat assessments were observed (25.4%; n = 32/126) and 17.5% (n = 22/126) for patients receiving eteplirsen at EVOLVE study entry. At least two repeat assessments were available for Brooke upper limb score/PUL 2.0 Entry Item A (66.7%; n = 84/126). A minimum of two repeat evaluations were identified in 61.9% (n = 78/126) of pulmonary FVC%p and 57.9% (n = 73/126) of cardiac LVEF clinical outcomes (Table 7).
Discussion
The EVOLVE observational, prospective study reports real-world data from the most extensive phase IV study of patients who are treated with PMO therapy to date. Using collated baseline patient characteristics, including functional data from EVOLVE, will help define the types of patients with DMD who are receiving PMO treatment in the real world.
The PMO body of evidence includes long-term post hoc analysis from clinical data suggesting PMOs have been associated with slowing DMD disease progression by delaying deterioration of LOA [25–27,43], pulmonary [27–29,44], cardiac function [31] and prolonging survival outcomes [32]. Additionally, the ESSENCE trial is a global, randomized, double-blind, placebo-controlled, phase III trial which evaluated patients ages 6–13 years old, with DMD amenable to exon 45 or 53 skipping for the efficacy and safety of casimersen and golodirsen compared with placebo. Topline results will be presented in 2026 and add to the growing body of PMO evidence. A real-world study of health resource utilization in eteplirsen-treated patients in the United States (n = 389) suggested a favorable effect of eteplirsen on DMD disease burden and progression, as measured by hospital encounters and days, emergency department visits and days, and pulmonary and cardiac management visits [45]. Further real-world and observational study data are needed to evaluate long-term commercial exposure to PMOs in patients with DMD [24–27].
Safety observations in this interim data report evaluating the EVOLVE study patient population are reflective of those previously reported in clinical trials. These functional and clinical data demonstrate the use of PMO therapies in a broader real-world patient population than has been previously demonstrated in clinical trials. Based on the timed function tests, ambulatory patients at EVOLVE study entry appeared to have advanced disease; however, baseline functional data values for rise from the floor and 10MWR appear to be valid and consistent with the literature [46,47]. Clinical manifestations of DMD such as cardiac and pulmonary symptoms measured by mean FVC%p and echocardiogram were reported in over half of patients at study entry. Mean FVC%p value was within the reasonable expected range of values reported in CINRG [48,49] and LVEF median was consistent with values reported in an untreated DMD population [50]. Given the similarities in pulmonary and cardiac data compared with historical cohorts, these baseline characteristics are reflective of patients with DMD.
In general, post-marketing data sources with robust clinical data collection across multiple centers for patients with DMD do not exist outside of structured data collection like the EVOLVE study. For example, physician-reported clinical outcomes are not reported in other large data sources for DMD such as claims data [51,52] and patient registries [53]. Patient-reported outcomes for DMD, such as health-related quality of life, are also absent in claims data and electronic health records [54].
The implementation of streamlined specific outcomes and EDC modules increased the robustness of data reporting in the EVOLVE study, evidenced by increased capture of upper limb data end points over the period from 2021 to 2023. The methods used in this study of reporting functional outcomes to assess real-world effectiveness of PMO therapies for patients with DMD help meet the objective of increasing our knowledge on the appropriateness of RWE data for evaluating treatment effectiveness. The EVOLVE protocol was amended to only include PUL 2.0 Entry Item A. PUL 2.0 Entry Item A can reliably be used for longitudinal assessment of upper limb function and progressive impairment in both ambulatory and nonambulatory patients with DMD [55]. An additional strength of this study is the flexibility to modify the study design as improved and novel assessments and advances in DMD therapy become available. Additional variables may be included in the data collection forms, leading to important insights into questions that were not originally anticipated.
Confounding factors of this study that do not affect treatment decisions include uncollected variables, uncaptured data and other forms of bias that influence the strength of causal inference in observational studies [56], including recall and immortal time bias [56,57]. Some characteristics, such as comedication, corticosteroid treatment or comorbidities, for example, may be potentially underreported and therefore not captured due to the observational nature of the study. Additionally, the lack of patient-reported outcome data from patients and caregivers is a limitation of this study and will be important to include in future analyses that investigate the impact of DMD therapies from the patient and caregiver perspectives in the real world. Outcomes assessed in the EVOLVE study are being conducted with routine care, may not be sufficiently comprehensive to assess clinical trial end points, and may be influenced by the composition of the study population in this descriptive interim analysis.
A large proportion of the study sample are patients who initiated PMO therapy prior to EVOLVE entry, and therefore, pre-treatment baseline data are not available for all participants. Additionally, because the EVOLVE study does not have an untreated group, any future comparative analysis would require independent data. Stratified results may be presented for specific subgroups of interest to explore the impact of this potential bias and/or effect measure modification.
Conclusion
This interim data report provides a descriptive evaluation of baseline patient characteristics, safety and treatment continuation to date in 161 patients receiving PMOs enrolled in the phase IV EVOLVE study, including 35 patients who were younger than 7 years at PMO initiation. The report also provides ambulation, upper limb, cardiac and respiratory functional baseline data from patients receiving eteplirsen. Real-world data derived from EVOLVE, the first and largest phase IV study of patients treated with PMOs to date, will continue to describe long-term clinical outcomes of patients with DMD.
Summary points
•
This interim data report describes the treatment patterns, safety and baseline functional assessments of phosphorodiamidate morpholino oligomer (PMO)-treated patients with Duchenne muscular dystrophy (DMD) from the ongoing real-world, phase IV, observational EVOLVE study.
•
Of the patients receiving PMOs, no treatment-emergent serious adverse events were found to be treatment related to date, which is consistent with previous clinical trial results.
•
Functional and clinical assessments collected at baseline included loss of ambulation, rise from the floor time, 10-meter walk/run time, Performance of Upper Limb Module for DMD 2.0, echocardiogram and forced vital capacity % predicted.
•
The number of patients continuing eteplirsen treatment to date was high (>90%), even among patients who were nonambulatory at or after initiation of eteplirsen.
•
These real-world data from EVOLVE, the first and largest phase IV study of patients treated with PMOs to date, will continue to describe long-term clinical outcomes of patients with DMD.
Author contributions
All authors contributed to protocol design, results interpretation and drafting of the manuscript.
Acknowledgments
The authors thank the patients, their families, and all investigators involved in this study. Ben Zola supported the operations of the EVOLVE study. Part of the material in this manuscript was presented at the 2024 Neuromuscular Study Group Annual Scientific Meeting held 20–22 September in Tarrytown, NY, USA. EVOLVE Study Group Investigators and Affiliations: Doris Leung (Kennedy Krieger Institute, Baltimore, MD, USA); Katherine Mathews (The University of Iowa, Iowa City, IA, USA); Leigh Ramos-Platt (Children's Hospital Los Angeles, Los Angeles, CA, USA); Farida Abid (Texas Children's Hospital, Houston, TX, USA); Mai ElMallah (Duke University Hospital, Durham, NC, USA); Ashutosh Kumar (Penn State Milton S. Hershey Medical Center, Hershey, PA, USA); Warren Marks (Cook Children's Medical Center, Fort Worth, TX, USA); Craig Zaidman (Washington University in St. Louis, St. Louis, MO, USA); Diana Bharucha-Goebel (Children's National Hospital, Washington, DC, USA); Barry Byrne (University of Florida, Gainesville, FL, USA); Megan Waldrop (Center for Gene Therapy, Nationwide Children's Hospital and Ohio State University Wexner Medical Center, Columbus, OH, USA); Robert Fryer (Columbia University Medical Center, New York, NY, USA); Michael Cartwright (Atrium Health Wake Forest Baptist Medical Center, Winston-Salem, NC, USA); Aravindhan Veerapandiyan (University of Arkansas for Medical Sciences, Arkansas Children's Hospital, Little Rock, AR, USA); Rebecca Scharf (University of Virginia Children's Hospital, Charlottesville, VA, USA); Hoda Abdel-Hamid (UPMC Children's Hospital of Pittsburgh, Pittsburgh, PA, USA); Cuixia Tian (Cincinnati Children's Hospital Medical Center & University of Cincinnati College of Medicine, Cincinnati, OH, USA); John Brandsema (Children's Hospital of Philadelphia, Philadelphia, PA, USA); Craig McDonald (University of California, Sacramento, CA, USA); Seth Perlman (Seattle Children's Hospital, Seattle, WA, USA); Gyula Acsadi (Connecticut Children's Medical Center, Farmington, CT, USA).
Financial disclosure
The study was funded by Sarepta Therapeutics, Inc. (MA, USA).
Competing interests disclosure
C Tian served as site principal investigator for the Sarepta EVOLVE study and an advisory board consultant for Sarepta Therapeutics, Inc. A Veerapandiyan received compensation for ad hoc advisory boards/consulting activity from AMO Pharma, AveXis, Biogen, Catalyst, Edgewise Therapeutics, Entrada, FibroGen, Italfarmaco, Lupin, Novartis, Pfizer, PTC Therapeutics, Sarepta Therapeutics, Inc., Scholar Rock, and UCB Pharma; receives research funding from AMO Pharma, Capricor Therapeutics, Edgewise Therapeutics, FibroGen, Muscular Dystrophy Association, Novartis, Parent Project Muscular Dystrophy, Pfizer, REGENXBIO, and Sarepta Therapeutics, Inc; and has other relationship(s) with MedLink Neurology for editorial services. RJ Scharf received research funding from argenx, AveXis/Novartis, Biohaven, Capricor Therapeutics, Genentech/Roche, and Sarepta Therapeutics, Inc. M Waldrop received research funding as site or study principal investigator from Alcyone Therapeutics, Inc., Novartis Gene Therapies, and Sarepta Therapeutics, Inc. and serves as a consultant for Sarepta Therapeutics, Inc. K Drummond, S Grabich, S Hornibrook, S Santra, and I Sehinovych are employees of Sarepta Therapeutics, Inc., and may own stock/options in the company. The authors have no other competing interests or relevant affiliations with any organization or entity with the subject matter or materials discussed in the manuscript apart from those disclosed.
Writing disclosure
Medical writing support was provided by Matthew Bidgood, PhD, and Kimberly Fischer, PhD of Eloquent Scientific Solutions, and was funded by Sarepta Therapeutics, Inc.
Ethical conduct of research
The authors state that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations. In addition, for investigations involving human subjects, informed consent has been obtained from the participants involved.
Data sharing statement
Qualified researchers may request access to the data that support the findings of this study from Sarepta Therapeutics, Inc., by contacting [email protected].
Open access
This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 Unported License. To view a copy of this license, visit https://creativecommons.org/licenses/by-nc-nd/4.0/
Supplementary Material
File (supplementary data.docx)
- Download
- 83.48 KB
References
1.
Bushby K, Finkel R, Birnkrant DJ et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 9(1), 77–93 (2010).
2.
Duan D, Goemans N, Takeda S, Mercuri E, Aartsma-Rus A. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 7(1), 13 (2021).
3.
Ricotti V, Ridout DA, Scott E et al. Long-term benefits and adverse effects of intermittent versus daily glucocorticoids in boys with Duchenne muscular dystrophy. J. Neurol. Neurosurg. Psychiatry 84(6), 698–705 (2013).
4.
Bello L, Morgenroth LP, Gordish-Dressman H, Hoffman EP, McDonald CM, Cirak S. DMD genotypes and loss of ambulation in the CINRG Duchenne Natural History Study. Neurology 87(4), 401–409 (2016).
5.
Birnkrant DJ, Bushby K, Bann CM et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 17(3), 251–267 (2018).
6.
Ryder S, Leadley RM, Armstrong N et al. The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: an evidence review. Orphanet J. Rare Dis. 12(1), 79 (2017).
7.
Mercuri E, Bönnemann CG, Muntoni F. Muscular dystrophies. Lancet 394(10213), 2025–2038 (2019).
8.
Lechner A, Herzig JJ, Kientsch JG et al. Cardiomyopathy as cause of death in Duchenne muscular dystrophy: a longitudinal observational study. ERJ Open Res. 9(5), 00176–02023 (2023).
9.
Landfeldt E, Thompson R, Sejersen T, McMillan HJ, Kirschner J, Lochmüller H. Life expectancy at birth in Duchenne muscular dystrophy: a systematic review and meta-analysis. Eur. J. Epidemiol. 35(7), 643–653 (2020).
10.
Nascimento Osorio A, Medina Cantillo J, Camacho Salas A, Madruga Garrido M, Vilchez Padilla JJ. Consensus on the diagnosis, treatment and follow-up of patients with Duchenne muscular dystrophy. Neurologia (Engl. Ed.) 34(7), 469–481 (2019).
11.
EXONDYS 51 (eteplirsen). Prescribing Information. Sarepta Therapeutics, Inc, MA, USA (2025).
12.
VYONDYS 53 (golodirsen). Prescribing Information. Sarepta Therapeutics, Inc, MA, USA (2024).
13.
VILTEPSO (viltolarsen). Prescribing Information. NS Pharma, Inc, NJ, USA (2021).
14.
AMONDYS 45 (casimersen). Prescribing Information. Sarepta Therapeutics, Inc, MA, USA (2024).
15.
ELEVIDYS (delandistrogene moxeparvovec-rokl). Prescribing Information. Sarepta Therapeutics, Inc, MA, USA (2024).
16.
DUVYZAT (givinostat). Prescribing Information. ITF Therapeutics, LLC, MA, USA (2024).
17.
EMFLAZA (deflazacort). Prescribing Information. PTC Therapeutics, Inc, NJ, USA (2024).
18.
AGAMREE (vamorolone). Prescribing Information. Catalyst Pharmaceuticals, Inc, FL, USA (2024).
19.
FDA. FDA Approves Nonsteroidal Treatment for Duchenne Muscular Dystrophy. (2024). Available at: https://www.fda.gov/news-events/press-announcements/fda-approves-nonsteroidal-treatment-duchenne-muscular-dystrophy
20.
GlobalNewswire. Catalyst Pharmaceuticals Reports FDA Approval of AGAMREE® (vamorolone) for Duchenne Muscular Dystrophy Granted to Santhera Pharmaceuticals. (2023). Available at: https://www.globenewswire.com/en/news-release/2023/10/26/2767947/13009/en/Catalyst-Pharmaceuticals-Reports-FDA-Approval-of-AGAMREE-vamorolone-for-Duchenne-Muscular-Dystrophy-Granted-to-Santhera-Pharmaceuticals.html
21.
FDA. FDA Expands Approval of Gene Therapy for Patients with Duchenne Muscular Dystrophy. (2024). Available at: https://www.fda.gov/news-events/press-announcements/fda-expands-approval-gene-therapy-patients-duchenne-muscular-dystrophy
22.
Mendell JR, Rodino-Klapac LR, Sahenk Z et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann. Neurol. 74(5), 637–647 (2013).
23.
Frank DE, Schnell FJ, Akana C et al. Increased dystrophin production with golodirsen in patients with Duchenne muscular dystrophy. Neurology 94(21), e2270–e2282 (2020).
24.
McDonald CM, Shieh PB, Abdel-Hamid HZ et al. Open-label evaluation of eteplirsen in patients with Duchenne muscular dystrophy amenable to exon 51 skipping: PROMOVI trial. J. Neuromuscul. Dis. 8(6), 989–1001 (2021).
25.
Mendell JR, Goemans N, Lowes LP et al. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann. Neurol. 79(2), 257–271 (2016).
26.
Mendell JR, Khan N, Sha N et al. Comparison of long-term ambulatory function in patients with Duchenne muscular dystrophy treated with eteplirsen and matched natural history controls. J. Neuromuscul. Dis. 8(4), 469–479 (2021).
27.
Mitelman O, Abdel-Hamid HZ, Byrne BJ et al. A combined prospective and retrospective comparison of long-term functional outcomes suggests delayed loss of ambulation and pulmonary decline with long-term eteplirsen treatment. J. Neuromuscul. Dis. 9(1), 39–52 (2022).
28.
Kinane TB, Mayer OH, Duda PW, Lowes LP, Moody SL, Mendell JR. Long-term pulmonary function in Duchenne muscular dystrophy: comparison of eteplirsen-treated patients to natural history. J. Neuromuscul. Dis. 5(1), 47–58 (2018).
29.
Iff J, Gerrits C, Zhong Y et al. Delays in pulmonary decline in eteplirsen-treated patients with Duchenne muscular dystrophy. Muscle Nerve 66(3), 262–269 (2022).
30.
Harper AD, Topaloglu H, Mercuri E et al. Safety and efficacy of viltolarsen in ambulatory and nonambulatory males with Duchenne muscular dystrophy. Sci. Rep. 14(1), 23488 (2024).
31.
Iff J, Desguerre I, Liu Y et al. Association between exon-skipping therapy with eteplirsen and cardiac outcomes in Duchenne muscular dystrophy. Presented at: MDA 2025 Clinical & Scientific Conference. TX, USA (March 16-19, 2025).
32.
Iff J, Done N, Tuttle E et al. Survival among patients receiving eteplirsen for up to 8 years for the treatment of Duchenne muscular dystrophy and contextualization with natural history controls. Muscle Nerve 70(1), 60–70 (2024).
33.
Wagner KR, Kuntz NL, Koenig E et al. Safety, tolerability, and pharmacokinetics of casimersen in patients with Duchenne muscular dystrophy amenable to exon 45 skipping: a randomized, double-blind, placebo-controlled, dose-titration trial. Muscle Nerve 64(3), 285–292 (2021).
34.
Mercuri E, Seferian AM, Servais L et al. Safety, tolerability and pharmacokinetics of eteplirsen in young boys aged 6–48 months with Duchenne muscular dystrophy amenable to exon 51 skipping. Neuromuscul. Disord. 33(6), 476–483 (2023).
35.
Iannaccone S, Phan H, Straub V et al. Casimersen in patients with Duchenne muscular dystrophy: interim safety and muscle biopsy results from the phase III ESSENCE trial. Presented at: 2023 MDA Clinical and Scientific Conference (2023).
36.
ClinicalTrials.gov. A long-term observational study evaluating eteplirsen, golodirsen, or casimersen in routine clinical practice (EVOLVE). (2019). Available at: https://clinicaltrials.gov/study/NCT06606340
37.
Ricchetti-Masterson K, Santra S, Hornibrook S et al. Interim analysis of EVOLVE: a long-term observational study evaluating eteplirsen, golodirsen, or casimersen in routine clinical practice. Presented at: 27th International Hybrid Annual Congress of the World Muscle Society. Halifax, NS, Canada (11–15 October 2022).
38.
Grabich S, Santra S, Waldrop MA et al. Interim analysis of EVOLVE: evaluating eteplirsen, golodirsen, or casimersen treatment in patients <7years old in routine clinical practice. Presented at: 28th International Annual Congress of the World Muscle Society. SC, USA (3–7 October 2023).
39.
Tian C, Veerapandiyan A, Grabich S et al. 2023 interim analysis of EVOLVE: a long-term observational phase IVstudy evaluating eteplirsen, golodirsen, or casimersen in routine clinical practice. Presented at: 2024 Neuromuscular Study Group Annual Scientific Meeting. NY, USA (20–22 September 2024).
40.
Waldrop MA, Grabich S, Santra S et al. Interim analysis of EVOLVE: evaluating eteplirsen treatment in nonambulatory patients in routine clinical practice from a phase IV observational study. Presented at: MDA Clinical and Scientific Conference. FL, USA (3–6 March 2024).
41.
McDonald CM. Timed function tests have withstood the test of time as clinically meaningful and responsive endpoints in duchenne muscular dystrophy. Muscle Nerve 58(5), 614–617 (2018).
42.
McDonald CM, Henricson EK, Abresch RT et al. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: a prospective cohort study. Lancet 391(10119), 451–461 (2018).
43.
Muntoni F, Seferian AM, Straub V et al. Six-year long-term safety and efficacy of golodirsen in patients with DMD vs mutation-matched external controls. Presented at: 28th International Annual Congress of the World Muscle Society. SC, USA (3–7 October 2023).
44.
Iff J, Tuttle E, Liu Y et al. Delayed pulmonary progression in golodirsen-treated patients with Duchenne muscular dystrophy vs mutation-matched external controls. Presented at: 28th International Annual Congress of the World Muscle Society. SC, USA (3–7 October 2023).
45.
Iff J, Zhong Y, Tuttle E, Gupta D, Paul X, Erik H. Real-world evidence of eteplirsen treatment effects in patients with Duchenne muscular dystrophy in the USA. J. Comp. Eff. Res. 12(9), e230086 (2023).
46.
Lott DJ, Taivassalo T, Senesac CR et al. Walking activity in a large cohort of boys with Duchenne muscular dystrophy. Muscle Nerve 63(2), 192–198 (2021).
47.
Stimpson G, Ridout D, Wolfe A et al. Quantifying variability in motor function in Duchenne muscular dystrophy: UK centiles for the NorthStar Ambulatory Assessment, 10 m walk run velocity and rise from floor velocity in GC treated boys. J. Neuromuscul. Dis. 11(1), 153–166 (2024).
48.
Khan N, Eliopoulos H, Han L et al. Eteplirsen treatment attenuates respiratory decline in ambulatory and non-ambulatory patients with Duchenne muscular dystrophy. J. Neuromuscul. Dis. 6(2), 213–225 (2019).
49.
Mayer OH, Finkel RS, Rummey C et al. Characterization of pulmonary function in Duchenne muscular dystrophy. Pediatr. Pulmonol. 50(5), 487–494 (2015).
50.
Awano H, Nambu Y, Itoh C et al. Longitudinal data of serum creatine kinase levels and motor, pulmonary, and cardiac functions in 337 patients with Duchenne muscular dystrophy. Muscle Nerve 69(5), 604–612 (2024).
51.
Szabo SM, Klimchak AC, Qian C, Iannaccone S, Popoff E, Gooch KL. Characterizing the occurrence of key clinical milestones in Duchenne muscular dystrophy in the United States using real-world data. J. Neuromuscul. Dis. 9(6), 689–699 (2022).
52.
Qian C, Klimchak AC, Szabo SM, Popoff E, Iannaccone ST, Gooch KL. Observing the clinical course of Duchenne muscular dystrophy in medicaid real-world healthcare data. Adv. Ther. 41(6), 2519–2530 (2024).
53.
Parent Project Muscular Dystrophy. Fifteen year registry report. (2023). Available at: https://www.parentprojectmd.org/wp-content/uploads/2023/08/PPMD_15-Year-Registry-Report_2023.pdf
54.
Gooch KL, Audhya I, Ricchetti-Masterson K, Szabo SM. Current challenges of using patient-level claims and electronic health record data for the longitudinal evaluation of Duchenne muscular dystrophy outcomes. Adv. Ther. 41(9), 3615–3632 (2024).
55.
Ayyar Gupta V, Pitchforth JM, Domingos J et al. Determining minimal clinically important differences in the North Star Ambulatory Assessment (NSAA) for patients with Duchenne muscular dystrophy. PLoS ONE 18(4), e0283669 (2023).
56.
Hammerton G, Munafò MR. Causal inference with observational data: the need for triangulation of evidence. Psychol. Med. 51(4), 563–578 (2021).
57.
Yadav K, Lewis RJ. Immortal time bias in observational studies. JAMA 325(7), 686–687 (2021).
Information & Authors
Information
Published In
Copyright
© 2026 The authors. This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 Unported License
History
Received: 28 June 2025
Accepted: 9 February 2026
Published online: 10 April 2026
Keywords:
Topics
Authors
Metrics & Citations
Metrics
Article Usage
Article usage data only available from February 2023. Historical article usage data, showing the number of article downloads, is available upon request.
Citations
How to Cite
Advancements from the EVOLVE study for assessing real-world experience with eteplirsen, golodirsen and casimersen for the treatment of DMD. (2026) Journal of Comparative Effectiveness Research. DOI: 10.57264/cer-2025-0108
Export citation
Select the citation format you wish to export for this article or chapter.
Citing Literature
- Sai Dharmarajan, Shannon Grabich, Richard Baxter, Aalok Nadkar, Carol Schermer, A Real-World Target Trial Emulation of Eteplirsen, Casimersen, and Golodirsen to Evaluate Survival Among Patients with Duchenne Muscular Dystrophy, Advances in Therapy, 10.1007/s12325-026-03609-0, (2026).