The efficacy and safety of GP40081 (insulin aspart biphasic 30) compared with NovoMix® 30 in Type 2 diabetes patients
Publication: Journal of Comparative Effectiveness Research
Abstract
Aim: To evaluate the safety and efficacy of insulin Aspart-Mix biosimilar candidate GP40081 (GP-Asp30) compared with NovoMix® 30 (NN-Asp30). Materials & methods: In a randomized open-label, active-controlled, 26-week non-inferiority clinical trial 264 patients with Type 2 diabetes mellitus were randomized 1:1 to receive once-daily GP-Asp30 or NN-Asp30. The primary safety end point was the immune response rate. Efficacy outcomes were a mean change in HbA1c (primary), frequency of achieving a glycemic g fasting plasma glucose levels, 7-point glucose profiles, and insulin doses. Results: The immune response developed in 10/126 (8%) participants in the GP-Asp30 group and in 10/125 (8%) participants in the NN-Asp30 group (p = 1.000). The mean difference in HbA1c change between groups was 0.12 (95%CI [-0.14, 0.38]). Other secondary efficacy and safety outcomes weren't statistically different between the two groups. Conclusion: GP-Asp30 demonstrated similar safety and efficacy compared with NN-Asp30 and may be considered a biosimilar insulin.
Worldwide healthcare systems face extra costs associated with an increasing number of diabetes patients treated with relatively expensive insulin analogues. Financial toxicity of diabetes treatment affects individual patients and national healthcare providers [1,2].
Biosimilars are defined as biological products that are highly similar to and have no clinically meaningful differences from an existing originator. Implementation of the biosimilarity concept into diabetes management can provide a decrease in insulin cost and improve its availability for patients [3].
Development of biosimilar drugs in general and insulin in particular is a strictly regulated stepwise process: analytical and in vitro studies, pharmacokinetics, and pharmacodynamics in human and immunogenicity trial [4,5].
Pre-mixed biphasic insulins contain short or rapid acting insulin combined with long or medium acting insulin. Although a basis-bolus insulin regimen gives more precise glycemic control, pre-mixed insulins may have the same efficacy and safety profile in appropriate patients [6].
Commonly, at the start of insulin therapy patients with Type 2 diabetes mellitus (T2D) are not candidates for the intensive basal/bolus regimen. Therefore, pre-mixed biphasic insulins seem to be a convenient and acceptable form for patients who are doing well on a stable, fixed ratio, particularly those who eat a larger breakfast and dinner and a smaller lunch, or are able to modify their diets to fit the kinetics of pre-mixed insulin [7,8].
In 2000 Novo Nordisk company registered biphasic insulin NovoMix 30® containing 30% soluble insulin aspart and 70% protamine-crystallized insulin aspart. The development of biphasic insulin is based on previously studied and registered insulin aspart substance.
GEROPHARM company developed and successfully registered insulin aspart GP40071 (GP-Asp) which is a biosimilar to Novo Nordisk referent. With GP-Asp as a proven biosimilar [9,10], a development of biphasic pre-mixed insulin aspart GP40081 (soluble insulin aspart/protamine-crystallized insulin aspart in the ratio 30/70) as a biosimilar to NovoMix 30® had started.
This clinical trial is the final step of the biosimilar insulin development program, and it aims to compare the safety (immunogenicity) and efficacy of GP-Asp30 and NN-Asp30. Since T2D patients are the target group of pre-mixed insulin adjustment, it is an appropriate way to study the immunogenicity of referent insulin and its biosimilar in this group.
Materials & methods
Study design & treatment
This was a phase III, multicenter, open-label, randomized, active-controlled, parallel-group, non-inferiority 26-week study conducted in 17 study sites from different regions of Russia. Eligible study participants with T2D (n = 264) in accordance with the inclusion/exclusion criteria were randomized 1:1 to GP-Asp30 (n = 132) or NN-Asp30 (n = 132) treatment.
The study consisted of 2 periods: (1) screening period – up to 4 weeks prior to intervention; (2) treatment period – 4 weeks of dose titration following 22 weeks of stable-dose treatment.
Through the dose titration period, insulin doses of both pre-mixed biphasic insulin and rapid-acting bolus insulin were selected and adapted to achieve glycemic control according to the established glycemic target [8,11]. During this period, oral antidiabetic drugs (OAD) could be switched or withdrawn and their doses could be changed as well.
During the following stable-dose treatment period, doses of pre-mixed insulin and OAD have been constant. The study participants self-adjusted bolus insulin consistently with their metabolic requirements during all study periods. To provide participants' safety during the stable-dose period pre-mixed insulin dose also could be changed by 15% (e.g., in cases of hypoglycemic episodes associated with dietary or physical activity changes or due to comorbidity), however, it was not recommended generally.
Study participants received insulin subcutaneously via self-injections with injector pens. The investigators taught study participants to administer self-injections and to calculate bolus insulin doses before insulin therapy had begun. To maintain stable blood glucose levels, participants did self-monitoring of blood glucose at least four-times a day.
The study was registered on ClinicalTrials.gov NCT04226105.
Study population
Eligible participants were 18 to 65 years old, diagnosed with T2D according to WHO criteria (1999–2013) [12] for at least 6 months before screening, with HbA1c level 7.6–12.0% and body mass index (BMI) 18.5–40.0 kg/m2 at screening. We included patients who were either insulin-naive or received insulin therapy at least 6 months prior to screening.
We included only T2D patients requiring insulin therapy, because they are considered to be a target group for pre-mixed biphasic insulin administration.
The key exclusion criteria are listed below:
•
Contraindications for pre-mixed biphasic insulin aspart treatment.
•
Severe insulin resistance with daily insulin requirement >1.5 IU/kg.
•
Change in basal or bolus insulin therapy (international non-proprietary name insulin) within 6 months before the screening.
•
History of severe hypoglycemia within the last 6 months before screening
•
Receiving GPP-1 receptors agonists.
•
Receiving any experimental medication or medical devices for 3 months prior to randomization.
•
Any vaccinations for 6 months prior to randomization.
•
Any acute inflammatory illnesses for 3 weeks prior to screening.
•
Regular administration of immunosuppressive drugs or/and immunomodulatory therapy.
Patients with diabetes complications in the advanced stage (proliferative diabetic retinopathy, severe peripheral diabetic neuropathy or autonomic neuropathy, diabetic nephropathy with estimated glomerular filtration rate <45 ml/min/1.73-m2, diabetic foot syndrome) found at screening or earlier were also not enrolled to the clinical trial.
The full list of the exclusion criteria is on ClinicalTrials.gov.
Immunogenicity
Immunogenicity outcome measures were the primary safety end points in this study. The primary immunogenicity end point was the frequency of immune response at week 26. Immune response was defined by the following criteria:
•
Anti-insulin antibody (AIA) concentration exceeding 10 IU/ml at week 26 for participants with a negative test at screening (AIA concentration of 10 IU/ml or less).
•
Increase in AIA concentration of more than 30% from baseline for subjects with AIA concentration more than 10 IU/ml at screening (positive test).
Secondary immunogenicity end points included:
•
Change in mean AIA concentration from baseline.
•
Presence of neutralizing AIA at week 26 in participants with negative test at baseline.
•
Frequency of developing clinically immune response at week 26.
AIA concentration was measured with a validated enzyme immunoassay method in the central laboratory [13]. To detect neutralizing AIA, we used iLiteTM Insulin Assay Ready Cells introducing the firefly luciferase reporter gene to their genome under the control of an insulin-dependent promoter. Neutralizing activity was established via binding of insulin alpha-chain and highly affinitive CD220 receptor in samples.
Clinically significant immune response was defined as an increase of ≥30% in AIA concentration from baseline concurrently with either increase of ≥0.2% in HbA1c from baseline or with an increase of 20% in insulin requirement compared with the stable dose.
Other safety end points
Other safety end points were incidence and severity of adverse events (AEs) including AEs of special interest (hypoglycemic events, ketoacidosis, injection site reactions, and hypersensitivity reactions). Each AE was assessed by the study investigators according to the following criteria: seriousness, severity, and relationship with the investigational or comparator insulin (WHO-UMC causality assessment).
All hypoglycemic events were documented by the patients in self-control diaries and then were transferred to electronic case report forms. The hypoglycemic event were defined as episodes of blood glucose ≤3.9 mmol/l) or as symptoms of hypoglycemia, need for assistance from other people, loss of consciousness, starting time of the hypoglycemia event, and nocturnal hypoglycemia were also considered to describe hypoglycemia episodes. Severe hypoglycemia was reported if a person needed any assistance from other people.
A serious AE (SAE) was any untoward medical occurrence at any drug dose that, in the opinion, either of the investigator or sponsor, resulted in death, was life-threatening, required hospitalization, led to persistent/significant disability/incapacity, or was congenital anomaly/birth defect. If an SAE occurred, it was reported to the sponsor within 24 hours. Every SAE was reviewed by the sponsor's central pharmacovigilance and medical teams based on their clinical judgment.
AE details were collected for all study participants from signing the informed consent form at their last visit. All non-serious AEs were periodically analyzed by the Sponsor's Medical team.
Efficacy end points
Efficacy end points were considered secondary and following the regulatory guidelines included an HbA1c change at week 26 from baseline (baseline defined at screening):
•
Percentage of participants who achieved HbA1c ≤7.0% at week 26.
•
Percentage of participants who achieved an individual glycemic target at week 26 based on HbA1c value.
•
Change in fasting plasma glucose (FPG) level at week 26 from baseline (baseline defined at screening).
•
Change in seven-point glucose profile (SPGP) at week 26 from baseline (baseline defined at the end of the titration period).
•
Change in daily insulin dose after week 22 from baseline (baseline defined at the end of the titration period).
•
Change in body weight at week 26 from baseline (baseline defined at screening).
•
Patients' satisfaction with the current diabetes treatment in Diabetes Treatment Satisfaction Questionnaire, status version (DTSQs) [14] and Diabetes Treatment Satisfaction Questionnaire, change version (DTSQc) [15] tests at week 26, DTSQs values were also compared with baseline ones (baseline defined at randomization).
HbA1c (in %) was measured in blood samples with an automated high-performance liquid chromatography method at screening and weeks 12, 16, 21 and 26. FPG level was measured in venous blood samples with an enzymatic UV test at screening, week 12 and 26. Both HbA1c and FPG were assessed in the central laboratory. SPGP measurements were performed by the participants with glucometers Accu-Chek Active in the weeks preceding planned visits.
DTSQs and DTSQc questionnaires measured patients' satisfaction with the current diabetes treatment. Patients completed DTSQs at randomization and week 26, while DTSQc was completed only at week 26.
Statistical analysis
An estimated sample size of 238 patients (119 participants per intervention group with GP-Asp30 and 119 per control with NN-Asp30) will provide >95% power to show non-inferiority of the GP‐Asp-Mix compared with NN‐Asp-Mix with respect to mean change in HbA1c (true difference of zero), non-inferiority upper margin of 0.4% (standard deviation [SD] of 1.1%; significance level of 2.5%, one‐sided t-test). Anticipating a 10% probability of early withdrawals, a total 264 would be randomized.
Statistical analyses were performed using Windows software version R 3.5.0 following the approved statistical analysis plan.
Results
Study participants
294 T2D patients were enrolled into the study. Among them, 264 were randomized to receive either GP-Asp30 (132) or NN-Asp30 (132). One participant from the NN-Asp30 arm was wrongly enrolled into the study and was excluded before the treatment period had begun. 263 participants received studied medications and were included in intention-to-treat (ITT) analysis.
255 participants completed the study at week 26: 127 and 128 participants received GP-Asp30 and NN-Asp30, respectively. During the treatment period, four participants decided to withdraw from the study, three participants were excluded by study protocol, and one participant was lost to follow-up. Totally 251 participants were included in the per-protocol (PP) analysis of immunogenicity. The flowchart of the study participants is shown in Figure 1.
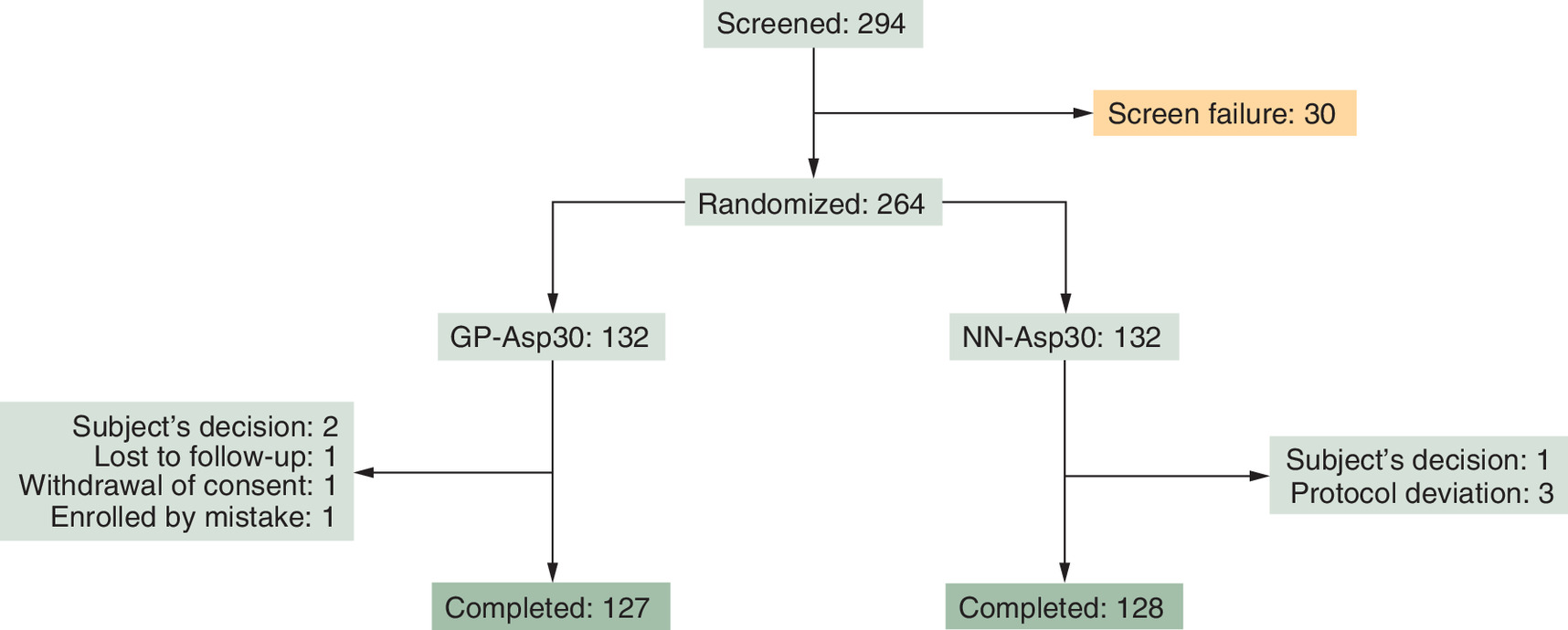
All participants who received at least 1 dose of any studied drug were included in the safety analysis.
All baseline characteristics were comparable between the two groups. Baseline characteristics of the study subjects are presented in Table 1.
| GP-Asp30 (n = 132) | NN-Asp30 (n = 131) | |
|---|---|---|
| Age, years | 57.4 ± 6.6 | 56.7 ± 7.3 |
| Gender (female) | 36 (27.3%) | 40 (30.5%) |
| Ethnicity (European) | 132 (100%) | 131 (100%) |
| Body weight, kg | 87.2 ± 14.3 | 87.7 ± 13.8 |
| BMI, kg/m2 | 31.4 ± 4.6 | 31.6 ± 4.5 |
| Smokers • Yes • No • Prior | 10 (7.6%) 116 (87.9%) 6 (4.5%) | 10 (7.6%) 114 (87.0%) 7 (5.3%) |
| Duration of diabetes, years | 11.5 ± 6.2 | 11.9 ± 7.1 |
| Subjects received insulin before trial started, n | 97 (73.5%) | 97 (74.1%) |
| Total insulin dose, IU/day | 38.3 ± 18.8 | 40.4 ± 19.1 |
| Basal insulin therapy | ||
| • Glargine 300 IU/ml • Glargine 100 IU/ml • Detemir • Degludec • Isophane | 11 (8.3%) 26 (19.7%) 6 (4.5%) 1 (0.8%) 7 (5.3%) | 15 (11.5%) 25 (19.1%) 8 (6.1%) 2 (1.5%) 3 (2.3%) |
| Basal insulin dose, IU/day | 24.5 ± 8.3 | 24.5 ± 10.0 |
| Bolus insulin therapy | ||
| • Aspart • Glulisine • Human soluble • Lispro | 1 (0.8%) 1 (0.8%) 6 (4.5%) 2 (1.5%) | 1 (0.8%) 1 (0.8%) 5 (3.8%) 0 (0%) |
| Bolus insulin dose, IU/day | 26.6 ± 8.2 | 26.2 ± 8.5 |
| Patients positive for AIA, n | 9 (6.8%) | 8 (6.1%) |
| Patients positive for neutralizing AIA, n | 6 (4.5%) | 7 (5.3%) |
| AIA concentration, IU/ml (at screening) | 4.79 ± 13.10 | 3.52 ± 6.60 |
| HbA1c,% | 9.18 ± 0.98 | 9.17 ± 0.99 |
| FPG, mmol/l | 11.81 ± 3.79 | 11.12 ± 3.52 |
| DTSQs, points | 21.34 ± 7.54 | 22.55 ± 7.36 |
Data for some baseline characteristics are mean ± standard deviation.
AIA: Anti-insulin antibodies; BMI: Body mass index; DTSQs: Diabetes Treatment Satisfaction Questionnaire (status version); FPG: Fasting plasma glucose; HbA1c: Glycated hemoglobin.
Immunogenicity
Immunogenicity was analyzed in the PP population with an additional subgroup analysis of insulin-naive patients and patients who received insulin therapy prior to our study.
At baseline, there were 9 (6.8%) AIA-positive patients in the GP-Asp30 group and 8 (6.1%) AIA-positive patients in the NN-Asp30 group. The frequency of immune response (according to criteria listed above) at week 26 did not differ between the two groups (p = 1.000): immune response developed in 10 of 126 (8%) participants in the GP-Asp30 group and in 10 of 125 (8%) participants in the NN-Asp30 group. Subgroup analysis showed no difference between insulin-naive patients and patients who previously received insulin in the frequency of immune response (p = 1.000).
There was no difference in baseline AIA concentration between the two groups (p = 0.991). The mean ± SD increase in AIA concentration at week 26 from baseline was 0.86 ± 6.39 IU/ml in the GP-Asp30 group (p = 0.001) and 2.29 ± 10.21 IU/ml in the NN-Asp30 group (p < 0.001). The change in AIA concentration did not differ statistically between the two groups (p = 0.587). Increasing in AIAs did not affect treatment efficacy (i.e., HbA1c level) according to post-hoc covariate analysis in ITT-population (p = 0.950).
Three (2.5%) of 121 subjects who were negative for neutralizing AIA at baseline became positive at week 26 in the GP-Asp30 group. And one (0.9%) of 117 subjects became positive for neutralizing AIA in the NN-Asp30 group.
The frequency of developing clinically significant immune response did not differ between the two groups independently on how it was defined (see above): either as an increase in AIA and in HbA1c or as an increase in AIA and in insulin dose. Results are presented in Table 2.
| ITT population | GP-Asp30 (n = 132) | NN-Asp mix (n = 131) | p-value |
|---|---|---|---|
| Participants with antibody positive and negative immune response at baseline, n (%) | |||
| Antibody positive | 9 (6.8) | 8 (6.1) | 1.000 |
| Antibody negative | 123 (93.2) | 123 (93.2) | |
| Participants with neutralizing AIAs, n, (%) | |||
| At screening At week 26 Newly developed | 6 (4.5) 3 (2.3) 0 (0) | 7 (5.3) 1 (0.8) 1 (0.8) | 0.989 |
| PP population | GP-Asp (n = 126) | NN-Asp (n = 125) | |
|---|---|---|---|
| AIA concentration. Data are mean ± standard deviation. | |||
| At baseline, IU/ml | 4.93 ± 13.39 | 3.59 ± 6.75 | 0.991 |
| At week 26, IU/ml | 5.79 ± 12.26 | 5.88 ± 11.21 | 0.587 |
| Frequency of immune response development based on AIA measurement at week 26, n, (%) | |||
| Patients with immune response Patients without immune response | 8 (9) 85 (89) | 8 (9) 83 (95) | 1.000 |
AIA: Anti-insulin antibody.
Safety
Safety was analyzed in all participants who received at least 1 dose of GP-Asp30 or NN-Asp30.
The incidence of AEs, SAEs, discontinuations due to AEs, and adverse drug reactions reported in the GP-Asp30 group were similar to those in the NN-Asp30 group (Table 3). Ten of 76 AEs in the GP-Asp30 group and seven of 55 AEs in the NN-Asp30 group were classified as adverse drug reactions, and their incidence did not differ between the groups (p = 0.412). No hypersensitivity reactions were observed in either of the groups. There were no differences in any laboratory safety values, vital signs and ECG between the groups.
| GP-Asp30 (n = 132) | NN-Asp30 (n = 131) | |
|---|---|---|
| Participants with one or more AEs, n, (%) | 46 (34.8) | 39 (29.8) |
| AEs (total), number of cases • mild • moderate • severe | 76 64 11 1 | 55 39 15 1 |
| Adverse drug reactions, number of cases | 9 | 6 |
| Serious AEs, number of cases | 5 | 3 |
| AEs of special interest, number of patients / number of cases • injection site reactions • hypersensitivity reactions | 1/13 0 | 0/0 0 |
AE: Adverse event.
There were 8 SAEs that were considered to be unrelated to drug exposure. All cases of SAE had good outcomes. There was no neither life-threatening AE nor lethal outcomes in our study.
Hypoglycemic episodes and injection site reactions were considered as AEs of special interest. Only one subject had an injection site reaction, the patient was in GP-Asp30 group. Table 4 contains the summary of hypoglycemic episodes. The number of patients experiencing at least one hypoglycemic episode did not differ between the groups (p = 1.000). Also, there was no difference in the overall number of hypoglycemic episodes (p = 0.080). Most patients in both groups experienced mild hypoglycemia.
| GP-Asp30 (n = 132) | NN-Asp30 (n = 131) | |||
|---|---|---|---|---|
| Subjects, n (%) | Events, n (incidence rate/patient-year) | Subjects, n (%) | Events, n (incidence rate/patient-year) | |
| Total patient years | 64.45 | 64.30 | ||
| Total | 58 (43.9) | 383 (5.9) | 57 (43.5) | 336 (5.2) |
| Severity | ||||
| • Mild | 58 (43.9) | 382 (5.9) | 57 (43.5) | 336 (5.2) |
| • Moderate | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| • Severe | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| • Unknown | 1 (0.8) | 1 (0.0) | 0 (0.0) | 0 (0.0) |
| Symptoms | ||||
| • Present | 51 (38.6) | 309 (4.8) | 51 (38.9) | 296 (4.6) |
| • Absent | 20 (15.2) | 72 (1.1) | 15 (11.5) | 35 (0.5) |
| • Unknown | 2 (1.5) | 2 (0.0) | 2 (1.5) | 5 (0.1) |
| Time of the episode | ||||
| • Night (00:00–05:59) | 19 (14.4) | 36 (0.6) | 15 (11.5) | 26 (0.4) |
| • Day (06:00–23:59) | 54 (40.9) | 347 (5.4) | 53 (40.5) | 310 (4.8) |
Efficacy
Here we provide efficacy outcomes analyzed in the ITT population, however, outcomes did not differ between PP and ITT populations.
At week 26 weeks of treatment, the observed mean ± SD HbA1c level decreased from 9.18 ± 0.98% at baseline to 8.25 ± 1.27% in GP-Asp30 and from 9.17 ± 0.99% at baseline to 8.12 ± 1.23% in NN-Asp30 (p < 0.001 for both). The following changes did not differ between the groups at week 26 (p = 0.416). The mean difference in HbA1c change with 95% CI at week 26 was 0.12 (-0.14, 0.38) – the confidence interval did not cross the established 0.4% non-inferiority margin demonstrating GP-Asp30 to be non-inferior to NN-Asp30 in terms of hypoglycemic efficacy. HbA1c changes are shown in Figure 2.
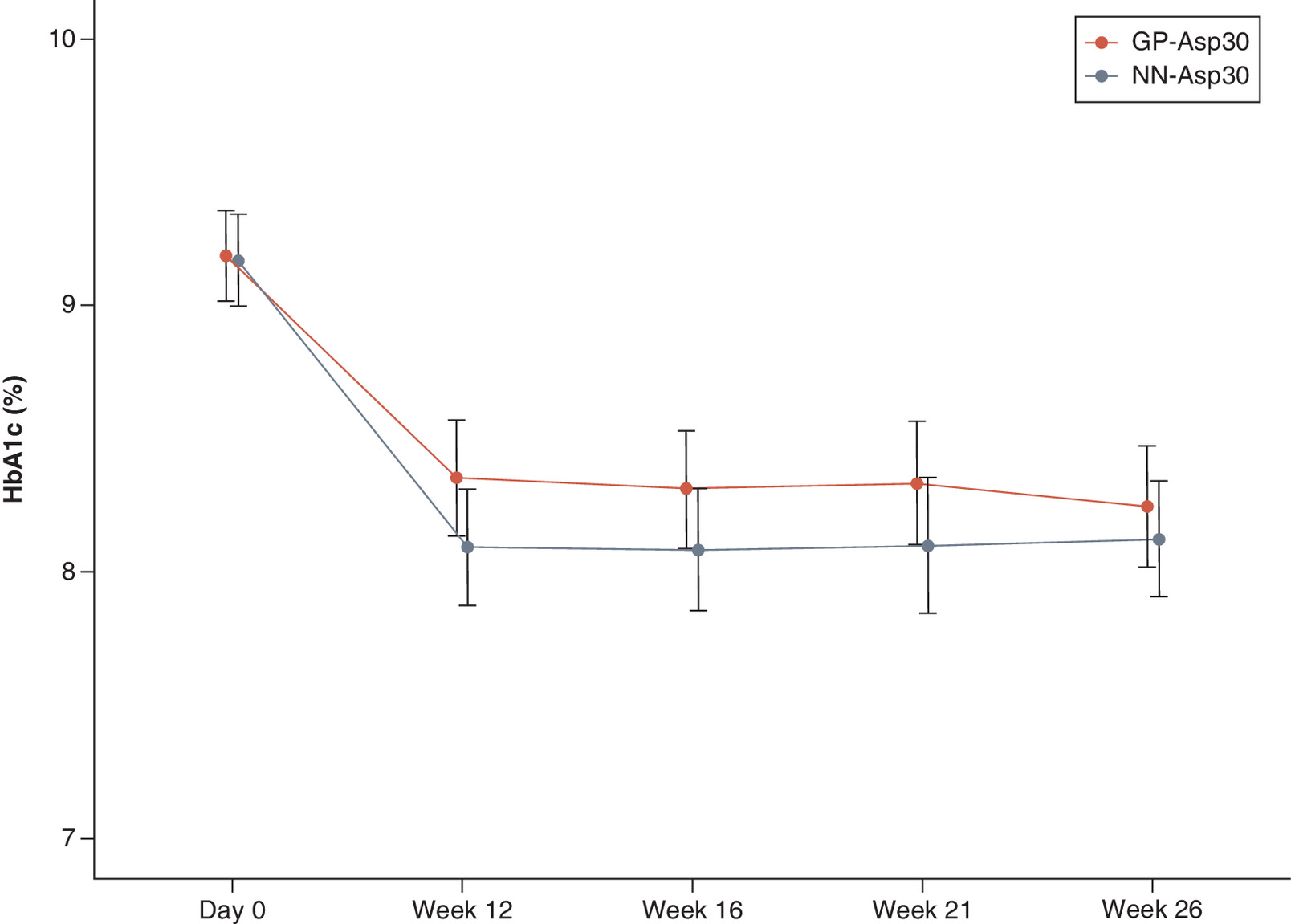
21 patients (16%) receiving GP-Asp30 and 25 patients (19%) receiving NN-Asp30 achieved an HbA1c level of ≤7.0%. The frequency of achieving an HbA1c level of ≤7.0% did not statistically differ between the two groups (p = 0.646). There was no significant difference in the rate of achieving individual glycemic targets as well (p = 0.624): 39 patients (30%) and 44 patients (34%) achieved their target HbA1c in GP-Asp30 and NN-Asp30 groups, respectively.
The observed mean ± SD FPG level significantly decreased from baseline at week 26 for 1.50 ± 4.55 mmol/l in the GP-Asp30 group (p < 0.001) and for 0.89 ± 4.51 mmol/l in the NN-Asp30 group (p = 0.008). The decrease in FPG levels did not differ between the two groups (p = 0.703). FPG level changes are shown in Figure 3.
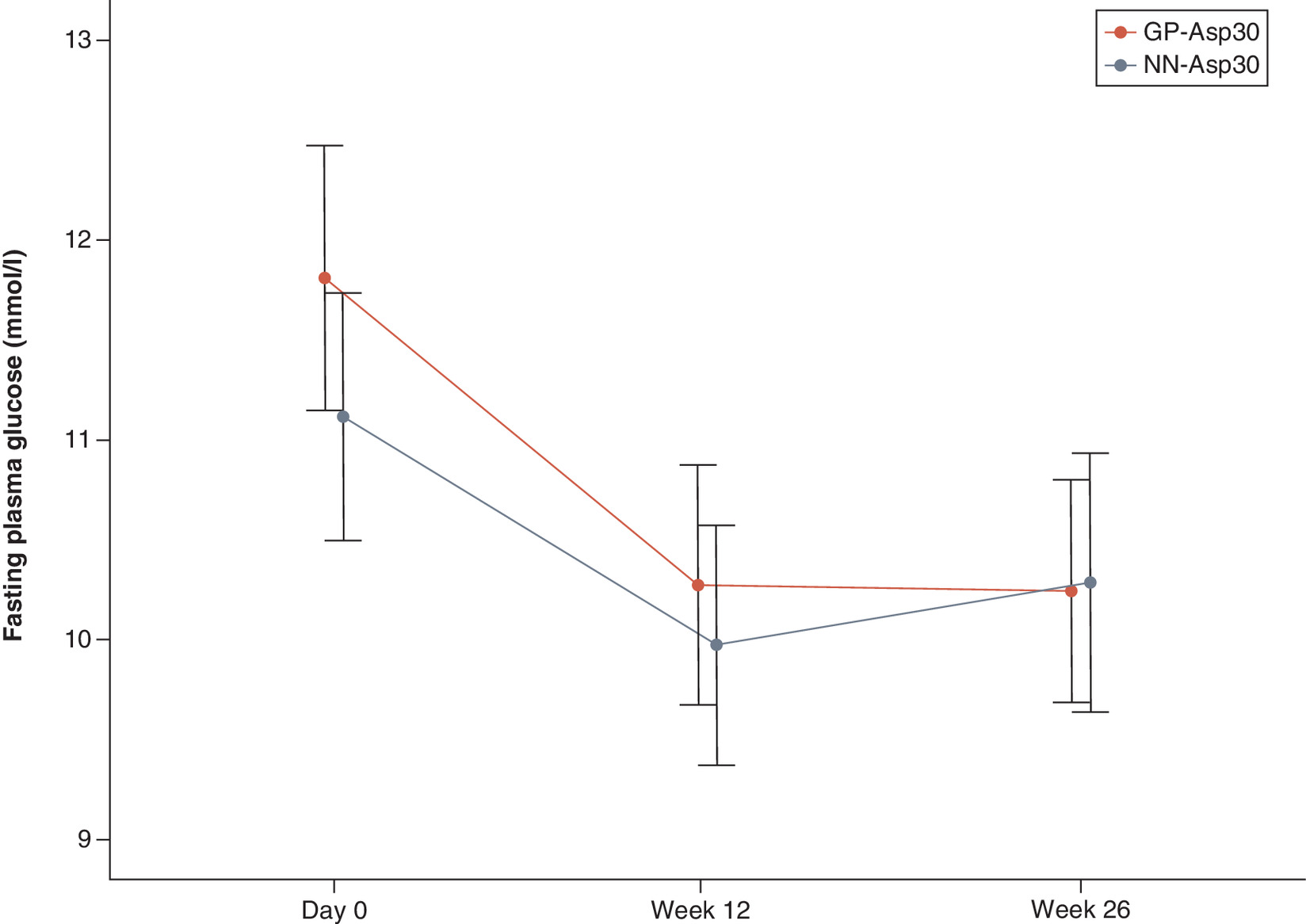
At week 26, mean changes in SPGP did not differ between the two groups in all measure points: before breakfast, 2 h after breakfast, before lunch, 2 h after lunch, before dinner, 2 h after dinner and 3 a.m. (p = 0.229; p = 0.835; p = 0.575; p = 0.428; p = 0.820; p = 0.683; p = 0.728, respectively). SPGP changes are shown in Figure 4.
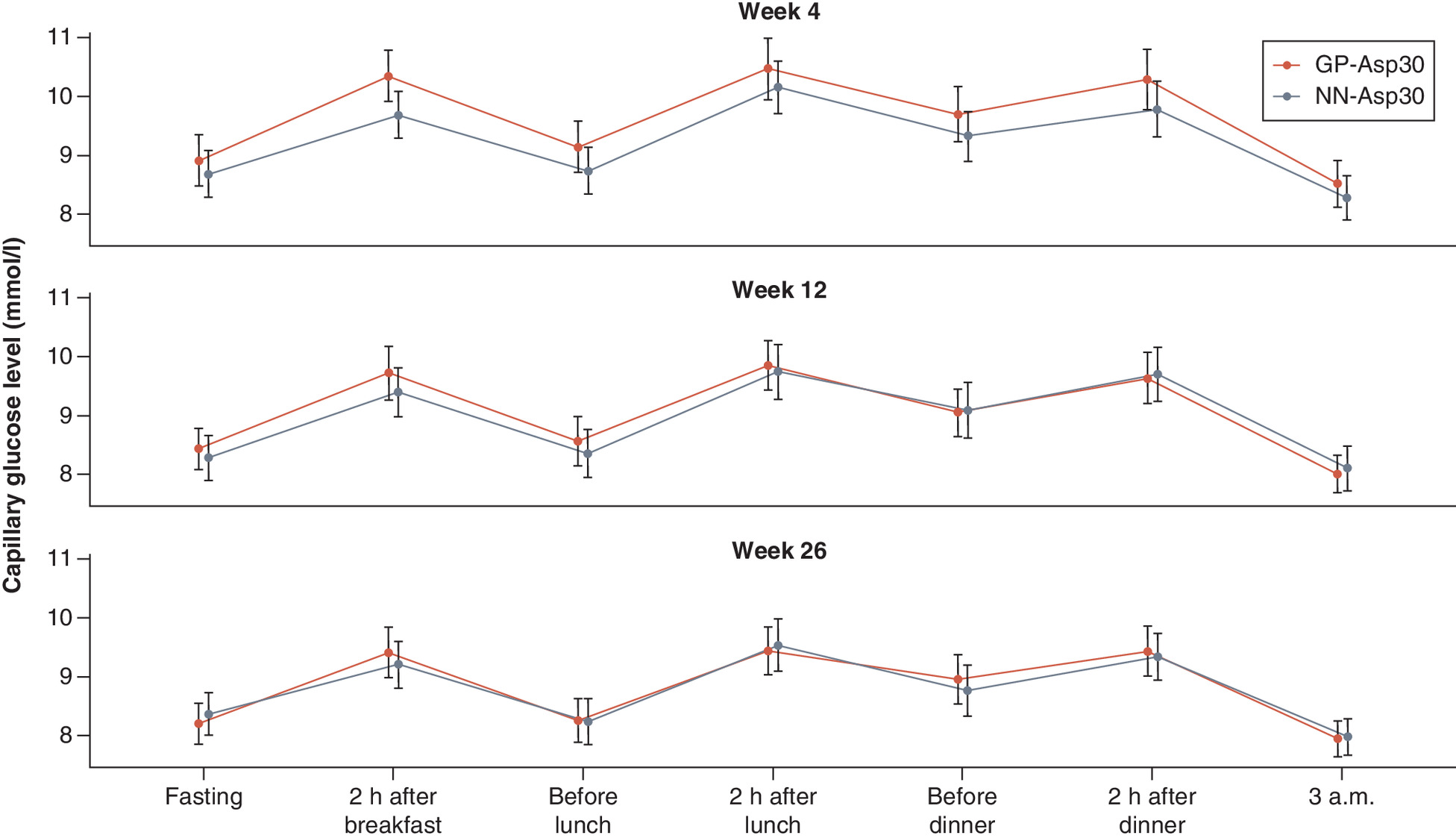
The mean ± SD daily insulin dosages increased at week 26 from the end of the titration period in both groups (p < 0.001 for both): 3.77 ± 5.38 IU/day in th GP-Asp30 group and 2.68 ± 4.08 IU/day in the NN-Asp30 group. There was no significant difference in the inter-group comparison of dosage changes at week 26 (p = 0.101).
At week 26 there was observed no change in body weight in GP-Asp30 group (-0.01 kg with 95% CI: [-0.39, 0.38]) and slight increase in NN-Asp30 group (0.21 kg with 95% CI: [-0.23, 0.65]). However, the difference between the two groups was insignificant (p = 0.478).
Treatment satisfaction at week 26 from baseline observed with DTSQs raised in both groups (p < 0.001) and did not statistically differ between groups (p = 0.081).
Discussion & recommendations
To prove biosimilarity of insulin product it is enough to show equivalence or non-inferiority compared with a referent in both pharmacodynamic/pharmacokinetic (PK/PD) profiles and to prove the same level of immunogenicity as referent has.
PK/PD profiles were studied earlier and no significant difference between GP-Asp30 and NN-Asp30 was shown.
Insulin immunogenicity is commonly assessed by measuring AIA levels, evaluation its neutralizing activity, and determining cases of clinically significant immune response.
In our controlled trial we showed GP-Asp30 to have a similar immunogenic profile compared with NN-Asp30 based on the frequency of immune response development as a primary end-point. Outcomes related to secondary immunogenicity end-points did not differ as well: change in AIA concentration, presence of its neutralizing activity, and rate of developing clinically immune response.
There were no significant differences in safety profiles including AEs and SAEs, between the groups received GP-Asp30 or NN-Asp30. Analysis of AEs of special interest such as hypoglycemic episodes and injection site reactions did not show any difference as well.
Our study showed GP-Asp30 to be non-inferior to the NN-Asp30, as measured by the change in HbA1c from baseline to week 26. Moreover, similar proportion of patients treated with GP-Asp30 and NN-Asp30 achieved either HbA1c ≤7% or their individual glycemic targets at week 26. Outcomes related to other secondary efficacy end-points were similar between the groups: FPG and SPGP levels, daily insulin dosages, change in body weight, and patients' satisfaction with the proposed treatment.
This study was a part of the GP-Asp30 (as a biosimilar to NN-Asp30) clinical development plan. Based on previous PK/PD studies and on the current results, GP-Asp30 has similar efficacy and safety (including immunogenicity) profiles. Finally, GP-Asp30 may be considered a biosimilar product since it successfully passed a confirmatory trial.
Future perspective
Biosimilar product development is a strictly regulated process aimed to prove biosimilar to have the same efficacy and safety as referent product. Biosimilar development programs cost less than original development. These two points make biosimilar development a good opportunity to increase accessibility to particular drugs.
Study limitations
This study has two main limitations: relatively small sample size and an open-label design.
There is no regulatory-approved or commonly recommended method of sample size estimation with respect to insulin immunogenic parameters. The sample size was estimated based on efficacy since it reflects the impact of probable immunogenic response to biosimilar insulin treatment. Even though the estimated sample size was sufficient to show non-inferiority for the efficacy end point, however, it was relatively small and it could be a limitation for analysis of other outcomes.
An open-label design could influence the investigators' approach to insulin dose titration. However it is not possible to blind such studies due to specific signs on insulin delivery systems and even is not required as immunogenicity end points are objective, and open-label design cannot influence them [4,5].
•
Diabetes care is affected by financial toxicity due to the relatively high cost of novel insulins and the biosimilarity concept can improve the availability of high-quality insulins for patients.
•
GP40081 (soluble insulin aspart)/protamine-crystallised insulin aspart in the ratio 30/70) is a biosimilar candidate for NovoMix 30® insulin and its' development is based on previously marketed biosimilar GP40071 (soluble insulin aspart).
•
GP40081 successfully passed the phase I trial (PK/PD study) previously and now it has completed phase III multicenter, open-label, randomized, active-controlled, parallel-group, non-inferiority trial.
•
This study aimed to compare the safety, immunogenicity and efficacy of investigated insulins.
•
A studied population of 264 T2D patients requiring insulin treatment received either GP40081 or NovoMix 30® through the total treatment period of 26 weeks.
•
Primary efficacy and immunogenicity outcomes were a mean change in HbA1c and a frequency of immune response at week 26, respectively.
•
The mean HbA1c change and the frequency of immune response at week 26 did not differ between groups as well as other secondary efficacy outcomes and safety analysis did not show any difference.
•
GP-Asp30 may be considered a biosimilar product since it successfully passed the confirmatory phase III trial.
Acknowledgments
The authors thank all investigational teams for taking part in this study. The authors also thank Unimed Laboratories and Quinta Analytica Yaroslavl for laboratory testing.
Financial & competing interests disclosure
This study was funded by GEROPHARM, Russia. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Ethical conduct of research
The trial was conducted in accordance with the Guidelines on Good Clinical Practice [16] and Ethical Principles for Medical Research Involving Human Subjects laid down in the Declaration of Helsinki [17]. All study participants provided written informed consent before participation in the study. The trial was approved by the Ethics Council (10 September 2019) and by the Ministry of Health of the Russian Federation (23 October 2019). The clinical study report was completed on 25 November 2020.
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
1.
Ong SE, Koh JJK, Toh SES et al. Assessing the influence of health systems on Type 2 diabetes mellitus awareness, treatment, adherence, and control: a systematic review. PLOS ONE 13(3), e0195086 (2018).
2.
Dedov II, Koncevaya AV, Shestakova MV et al. Economic evaluation of Type 2 diabetes mellitus burden and its main cardiovascular complications in the Russian Federation. Diabetes Mellitus 19(6), 518–527 (2016).
3.
White J, Goldman J. Biosimilar and follow-on insulin: the ins, outs, and interchangeability. J. Pharm. Technol. 35(1), 25–35 (2019).
4.
European Medical Agency. Guideline on non-clinical and clinical development of similar biological medicinal products containing recombinant human insulin and insulin analogues. MEA/CHMP/BMWP/32775/2005_Rev. 1 (2015).
• Regulatory guidelines that were fundamentally important for clinical development planning in general and for designing our phase III trial in particular. Primary outcome selection, sample size estimation, statistical analysis and other design considerations were made in a strong accordance with these guidelines.
5.
Eurasian Economic Union. Resolution No. 89 of November 3, 2016 On Approval of the Rules for Conduct of Studies of Biological medicines in the, Chapter 15.7 Preclinical and clinical development of bioanalogue (biosimilar) medicinal products containing recombinant insulin and insulin analogues. (2016).
• Regulatory guidelines that were fundamentally important for clinical development planning in general and for designing our phase III trial in particular. Primary outcome selection, sample size estimation, statistical analysis and other design considerations were made in a strong accordance with these guidelines.
6.
Zaykov A, Mayer J, DiMarchi R. Pursuit of a perfect insulin. Nat. Rev. Drug Discov. 15, 425–439 (2016).
7.
American Diabetes Association. 9. Pharmacologic approaches to glycemic treatment: standards of medical care in diabetes – 2021. Diabetes Care 44(S1), 111–124 (2021).
8.
Dedov II, Shestakova MV, Mayorov AY et al. Standards of specialized diabetes care (9th ed.). Diabetes Mellitus 22(S1), 1–144 (2019).
•• This publication was one of the main diabetes treatment guidelines used in population formation, dosing regimens (especially in dose titration period), etc.
9.
Drai RV, Karonova TL, Mayorov AY et al. Clinical pharmacology of insulin aspart biosimilar GP40071: pharmacokinetic/pharmacodynamic comparability in hyperinsulinemic euglycemic clamp procedure. Clin. Pharmacol. Drug Development. 11(8), 922–929 (2022).
•• This publication emphasizes the origin of GP40081 development and describe similar clinical development of related GP40071 insulin biosimilar. There are some methodological similarities between development programs and phase III trial design.
10.
Karonova TL, Mayorov AY, Magruk MA et al. Safety and efficacy of GP40071 compared with originator insulin aspart (NovoRapid® Penfill®) in Type 1 diabetes mellitus. J. Comp. Eff. Res. 10(9), 763–775 (2021).
•• This publication emphasizes the origin of GP40081 development and describe similar clinical development of related GP40071 insulin biosimilar. There are some methodological similarities between development programs and phase III trial design.
11.
American Diabetes Association. 6. Glycemic targets: standards of medical care in diabetes – 2021. Diabetes Care 44(S1), 111–124 (2021).
•• This publication was one of the main diabetes treatment guidelines used in population formation, dosing regimens (especially in dose titration period), etc.
12.
World Health Organization. Definition, diagnosis and classification of diabetes mellitus and its complications: report of a WHO consultation. Part 1, Diagnosis and classification of diabetes mellitus. No. WHO/NCD/NCS/99.2 (1999).
13.
Abramenko NB, Vnukova PI, Golovina ES et al. Development and validation of approach for the detection of neutralizing antibodies against insulin (glargine) in human blood plasma. Drug Dev. Res. 8(3), 70–78 (2019).
• This publication comprehensively describes diagnostic test system used in immunogenicity assessment.
14.
Bradley C, Lewis KS. Measures of psychological well-being and treatment satisfaction developed from the responses of people with tablet-treated diabetes. Diabet. Med. 7, 445–451 (1990).
15.
Bradley C. The Diabetes Treatment Satisfaction Questionnaire (DTSQ): change version for use alongside status version provides appropriate solution where ceiling effects occur. Diabetes Care 2(3), 530–532 (1999).
16.
Bhatt A. International Council for Harmonisation E6 (R2) addendum: challenges of implementation. Persp. Clin. Res. 8(4), 162 (2017).
17.
World Medical Association. World Medical Association Declaration of Helsinki. Ethical principles for medical research involving human subjects. Bull. World Health Organ. 79(4), 373 (2001).
Information & Authors
Information
Published In
Pages: 1337 - 1347
PubMed: 36511777
Copyright
© 2022 Future Medicine Ltd.
History
Received: 27 September 2021
Accepted: 14 October 2022
Published online: 13 December 2022
Keywords:
Topics
Authors
Funding Information
GEROPHARM, Russia
Metrics & Citations
Metrics
Article Usage
Article usage data only available from February 2023. Historical article usage data, showing the number of article downloads, is available upon request.
Citations
How to Cite
The efficacy and safety of GP40081 (insulin aspart biphasic 30) compared with NovoMix® 30 in Type 2 diabetes patients. (2022) Journal of Comparative Effectiveness Research. DOI: 10.2217/cer-2021-0232
Export citation
Select the citation format you wish to export for this article or chapter.
Citing Literature
- Xiaoxuan Xing, Lingyi Zhao, Ke Wang, Zhizhou Wang, Lan Zhang, Xianzhe Dong, Therapeutic equivalence and switching between biosimilar and reference insulins: A systematic review and meta‐analysis of randomised controlled trials, Diabetes, Obesity and Metabolism, 10.1111/dom.70328, 28, 2, (1371-1382), (2025).
- Ekaterina Koksharova, Roman Drai, Sergei Noskov, Artem Dorotenko, Ekaterina Protsenko, Kseniia Radaeva, Anna Arefeva, Maria Gefen, Gagik Galstyan, Igor Makarenko, Clinical Pharmacology of GP40321 (Insulin Glulisine Biosimilar): Pharmacokinetic and Pharmacodynamic Comparability in a Hyperinsulinemic‐Euglycemic Clamp Procedure, Clinical Pharmacology in Drug Development, 10.1002/cpdd.1401, 13, 7, (828-836), (2024).
- Thomas Danne, Lutz Heinemann, Thomas R. Pieber, New Insulins, Biosimilars, and Insulin Therapy, Diabetes Technology & Therapeutics, 10.1089/dia.2024.2504, 26, S1, (S-45-S-67), (2024).