Efficacy and safety of GP40021 insulin lispro biphasic compared with Humalog Mix 25 in Type 2 diabetes mellitus patients
Publication: Journal of Comparative Effectiveness Research
Abstract
Aim: To compare safety (immunogenicity) and efficacy of a biosimilar insulin GP-Lis25 and a reference insulin Ly-Lis25 (Humalog Mix 25) in Type 2 diabetes mellitus (T2D) patients. Materials & methods: This randomized open-label, 26-week clinical trial enrolled 210 T2D patients, randomized 1:1 to twice-daily GP-Lis25 or Ly-Lis25. The primary end point was immune response at 26th week. Noninferiority margin for HbA1c was 0.4%. Results: Immune response frequency was similar in GP-Lis25 and Ly-Lis25 groups both at week 12 (p = 0.651) and 26 (p = 0.164). The difference of HbA1c change at week 26 was (95% CI) 0.01 (-0.27–0.28)%. Fasting plasma glucose, seven-point glucose profile and insulin dose were similar between groups. Safety did not differ between groups. Conclusion: GP-Lis25 and Ly-Lis25 demonstrated similar safety and efficacy.
ClincalTrials.gov identifier: NCT04023344.
Diabetes mellitus (DM) is a serious problem worldwide which is associated with enormous economic losses. Insulin therapy is the only method for treatment of Type 1 DM (T1D) and one of the most important options for Type 2 DM (T2D) therapy. Insulin treatment allows to achieve stable glycemic control thereby preventing or slowing down the progression of DM complications and improving quality of life of patients [1].
Since insulin is a biological drug, its products, especially insulin analogues, are rather expensive and burdensome for state economics. A biosimilar is a biological medicinal product highly similar to another already approved biological medicinal product (the ‘reference medicine’) that is as safe and effective as the originator [2]. Being equally effective as the reference products, biosimilar medicinal products have many economic benefits allowing to increase accessibility of insulin analogues [3]. Therefore, introduction of the biosimilars to the market allows a patient and state economics to choose between different suppliers of the same medicinal product and reduce treatment cost significantly when choosing a biosimilar.
Insulin lispro biphasic 25 is a mixture of rapid-acting insulin lispro (25%) and insulin lispro protamine suspension with prolonged effect (75%). The originator Humalog Mix 25 (Ly-Lis25) is produced by Eli Lilly and Co (Fegersheim, France). Insulin lispro biphasic 25 (GP40021 [GP-Lis25]) was developed as a biosimilar to Ly-Lis25.
We have performed the development program using two drugs with the same active ingredient: insulin lispro (GP-Lis) and insulin lispro biphasic 25 (GP-Lis25) in accordance with the regulatory guidelines of the Eurasian Economic Union and European Medical Agency [4,5].
The similarity of GP-Lis25 to Ly-Lis25 and GP-Lis to Humalog was already proved on the basis of the head-to-head analytical and preclinical comparability studies. The euglycemic clamp studies were conducted both for GP-Lis (NCT03604575) and GP-Lis25 (NCT03606018). These studies demonstrated a high degree of similarity between GP-Lis and Humalog [6], and between GP-Lis25 and Ly-Lis25 [7].
A comparative safety (immunogenicity) and efficacy study of insulin biosimilar and reference drug is the last stage of the development program and it should be considered as a confirmatory not the pivotal study. If a biosimilar manufacturer develops different preparations containing the same active ingredient, the drug with the highest expected immunogenic potential should be included in the safety study [4,5]. GP-Lis25 is expected to be more immunogenic compared with GP-Lis as a fraction of protamine suspension contains protamine sulphate. The study results can be extrapolated to GP-Lis as a less immunogenic insulin preparation.
This clinical trial was conducted to compare safety (immunogenicity) and efficacy of GP-Lis25 and Ly-Lis25 and confirm their similarity.
Materials & methods
Study design & treatment
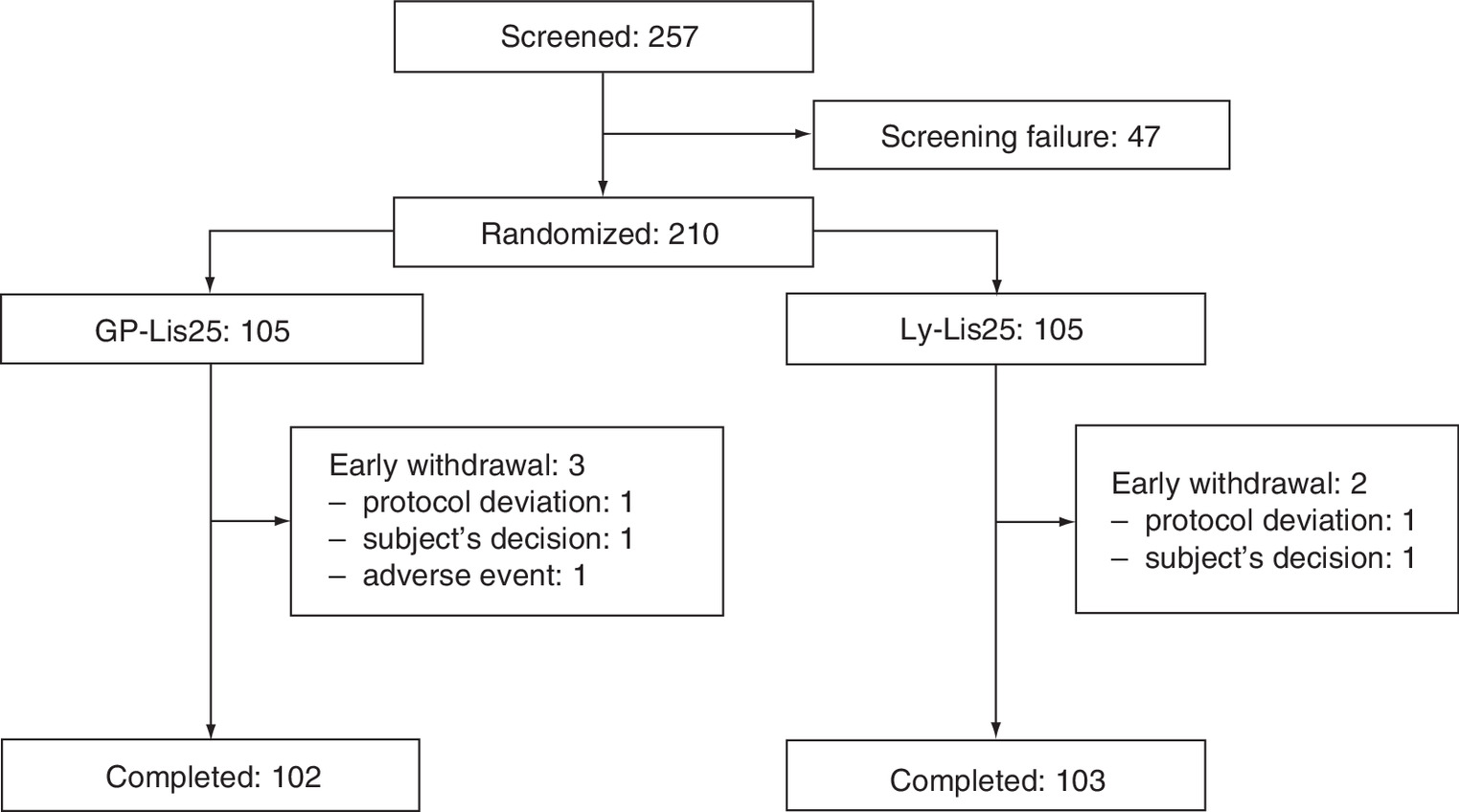
This was a Phase III, multicenter, open-label, randomized, active-controlled, parallel group, noninferiority 26-week study in 18 study sites Supplementary Material 1. Eligible study participants with T2D (n = 210) in accordance with the inclusion/exclusion criteria were randomized 1:1 to GP-Lis25 (n = 105) or Ly-Lis25 (n = 105) treatment twice-daily. We performed block randomization with the consideration of HbA1c level (7.6–9.0% or >9.0%) and previous insulin treatment (insulin-naive or currently treated with insulin). Figure 1 shows the flowchart of study subjects.
The study period consisted of screening procedure (up to 4 weeks), insulin dose titration (up to 4 weeks) and treatment period with stable doses (26 weeks). Study participants used prefilled insulin pens. During the period of insulin dose titration, insulin doses could be changed as much as needed to achieve optimal glycemic control in patients. Daily insulin doses during the treatment period should be stable. In special cases (e.g., to provide a subject’s safety if he has frequent hypoglycemic episodes), investigators could reduce insulin dose even during the treatment period with stable doses, however it was not recommended generally. To maintain stable blood glucose levels, participants should measure capillary blood glucose at least four-times a day, and every time they were supposed to have hypoglycemic episode.
The study was registered under ClinicalTrials.gov number NCT04023344.
Study population
Eligible participants were 18 to 65 years old, with T2D diagnosed for at least 6 months before screening, with HbA1c level 7.6–12.0% and BMI 18.5–35.0 kg/m2 at screening.
Participants regularly taking oral antidiabetic drugs before screening should continue administration in the same dose for at least 3 months before the first dosing of the investigational insulin. However, study subjects could discontinue sulphonylurea treatment in titration period in case of high hypoglycemic risk. Investigators made a decision based on a patient’s glycemic profile.
Only T2D patients took part in the trial as T1D patients had the increased risk of DM decompensation associated with nonpriority indication of insulin mixture. This waiver from the regulatory guidelines [4,5] was approved by the Ministry of Health of the Russian Federation.
Key exclusion criteria were the following:
•
anti-insulin antibody (AIA) level >10 units/ml;
•
insulin resistance (daily insulin requirement >1.5 units/kg);
•
use of three or more different oral hypoglycemic drugs;
•
history of hypoglycemia associated with seizure or loss of consciousness within the last 6 months;
•
history of ketoacidosis or treatment of uncontrolled DM within the last 6 months;
•
regular administration of immunosuppressive drugs or/and immunomodulatory therapy.
The full list of the exclusion criteria is provided at ClinicalTrials.gov.
Immunogenicity outcome measures
Immunogenicity outcome measures were the main safety end points in this study.
The primary end point was frequency of immune response (AIA concentration >10 units/ml) at week 26.
Secondary immunogenicity end points included mean AIA concentration at weeks 12 and 26, and the change in mean AIA concentration from baseline (at screening) to weeks 12 and 26.
Blood samples of patients having immune response either at week 12, or at week 26, or both, and a more than fourfold increase of AIA concentration at week 12, or at week 26, or both were tested for insulin neutralizing antibodies.
AIA concentration was assessed using the validated enzyme immunoassay method in the central laboratory.
To detect insulin neutralizing antibodies, we used the method with iLiteTM Insulin Assay Ready Cells, introducing the firefly luciferase reporter gene to their genome under the control of an insulin-dependent promoter [8]. Neutralizing activity was established via binding of insulin alfa-chain and CD220 receptor in samples.
Efficacy end points
Efficacy end points were considered as secondary in accordance with the regulatory guidelines [4,5] and included:
•
HbA1c change at week 26 from baseline (at screening),
•
change in fasting plasma glucose (FPG) level at week 26 from baseline (at screening),
•
change in seven-point glucose profile (SPGP) at week 26 from baseline (in the end of titration period – week 4),
•
change in insulin dose at week 26 from baseline (in the end of titration period – week 4),
•
change in body weight at week 26 from baseline (at screening),
•
an adjustment of patients’ satisfaction with current diabetes treatment in Diabetes Treatment Satisfaction Questionnaire – status (DTSQs) and Diabetes Treatment Satisfaction Questionnaire – change (DTSQc) tests at week 26, DTSQs were also compared with baseline values (at randomization).
The study investigators selected individual glycemic goal for each participant in accordance with the national guidelines on diabetes treatment [9] at the randomization visit. They did not change this goal during the study periods. HbA1c was measured at screening and at weeks 12 and 26.
FPG level was measured at the screening, in the beginning of treatment, at week 12 and 26. Glycemic level was also controlled daily by the study subjects at least four-times a day using glucometers given by Sponsor at the screening.
SPGP was performed by the participants using glucometers Accu-Chek Active with appropriate testing strips provided by the Sponsor. The study subjects performed SPGP three days a week before visits at weeks 4, 12 and 26, twice in two consecutive days. They measured capillary blood glucose in fasting condition, two hours after breakfast, before lunch, two hours after lunch, before dinner, two hours after dinner and at 3 a.m.
DTSQs and DTSQc questionnaires measure patients’ satisfaction with current diabetes treatment. Both questionnaires consist of eight questions estimating absolute and relevant satisfaction values for DTSQs and DTSQc, respectively. Patients completed DTSQs at randomization and at week 26, DTSQc – only at week 26.
Safety end points
In the study, incidence and severity of adverse events (AEs) including AEs of special interest, such as hypoglycemic episodes, injection site reactions and hypersensitivity reactions, were assessed. In addition to these secondary safety end points, changes in vital signs, changes in electrocardiogram and laboratory results compared with baseline, were also assessed. All investigators should report both all AEs observed in the study and AEs spontaneously reported by study subjects. AE details were collected for all study participants from signing the Informed Consent Form to their last visit.
Each AE was assessed by the study investigators according to the following criteria: seriousness, severity, relationship to the investigational product or comparator (WHO-UMC causality assessment). All AEs were also recorded in source documents and electronic case report forms. If a serious adverse event (SAEs) occurred, it was reported to the sponsor within 24 h. Every SAE was reviewed by the Sponsor’s central Pharmacovigilance and Medical teams based on their clinical judgment. All nonserious AEs were periodically analyzed by the Sponsor’s Medical team.
Hypoglycemic episodes, allergic reactions and injection site reactions were AEs of special interest (according to the study protocol), and they were evaluated separately from other AEs. Hypoglycemic episodes were recorded by the participants in their personal self-care glycemic diaries. Only episodes with capillary blood glucose level less than 3.9 mmol/l were considered hypoglycemic episodes. Physicians assessed hypoglycemia severity depending on a clinical situation, regardless the need in assistance of other people (in the same way they evaluated severity of any AEs). This fact was recorded separately. Minor and major episodes were also registered based on the need of external help. Local injection site reactions and hypersensitivity reactions were assessed during visits of every patient to the study site and were also recorded by the study participants in their diaries. The data were entered into electronic case report forms.
All these measures were used for provision of continuous benefit-risk assessment during the study.
Statistical analysis
All data analyses were made using R version 3.5.0 for Windows software in accordance with the approved statistical analysis plan. The results of statistical analysis in full analysis set population were presented.
Quantitative statistical analysis is described using:
•
quantitative observations (n);
•
mean variables (mean);
•
95% CI;
•
standard deviations;
•
median (median);
•
the lowest and highest quintile (Q1, Q3);
•
minimum and maximum values (min, max).
We used analysis of covariance (ANCOVA) to estimate changes of continuous variables from baseline. For each of these variables, ANCOVA included the baseline variable value as a covariate and treatment group and study site as fixed factors. Student’s t-test or Mann–Whitney U-test (depending on normality of distribution) were used to compare absolute values of continuous variables between the study groups. If variance was not equal in the groups (evaluated with the Levene’s test), Welch’s unequal variances t-test was used instead of Student’s t-test.
Qualitative statistical analysis is described using:
•
absolute frequency (quantitative observations);
•
relative frequency (%).
We compared qualitative (categorical) variables using X2-test (with continuity correction in case any unit contains value of 6 to 10) or exact Fisher’s test (if any unit contained value of 0 to 5). We analyzed the change of dichotomous variables from baseline with McNemar test. p-value < 0.05 was accepted statistically significant.
Sample size estimation
Since insulin immunogenicity is relatively low, high study power is not required for estimation of frequency of immune response [4,5]. In comparative studies of insulin immunogenicity, sample size is estimated on the basis of HbA1c values in accordance with the regulatory guideline [4,5]. We used α level 0.05, β level 0.2, mean difference 0, standard deviation 1.1% and δ 0.4% in accordance with the regular practice for antihyperglycemic therapy [10]. With these inputs, the total number of required study subjects was estimated as 186 (93 per group). Taking into account a possible 10–15% early withdrawal rate, we randomized 210 patients.
Regulatory & independent ethics committee approval
The trial was conducted in accordance with the guidelines on Good Clinical Practice and with ethical principles for medical research involving human subjects laid down in the Declaration of Helsinki [11]. All study participants provided the written informed consent before participating in the study. The trial was approved by the Ministry of Health of the Russian federation (Clinical trial authorization No. 644 dated 11 December 2017) and by the Ethics Council (abstract from the meeting minutes No 159 dated 21 December 2017).
Results
Study participants
We screened 257 T2D patients and randomized 210 of them (105 to GP-Lis25, 105 to Ly-Lis25 groups). Three participants discontinued GP-Lis25 therapy (one subject decided to withdraw from the study, another one was lost to follow-up, the third one had adverse reactions), and two of the subjects discontinued Ly-Lis25 therapy (one subject decided to withdraw from the study, another one was lost to follow-up). Figure 1 provides the flowchart of the study participants. Baseline characteristics of the study subjects are presented in Table 1. All baseline characteristics were comparable between the groups except prior insulin therapy: more patients in Ly-Lis25 group were administered with only basal insulin at baseline.
| Characteristics | GP-Lis25 (n = 105) | Ly-Lis25 (n = 105) | p-value |
|---|---|---|---|
| Age, years | 57.37 ± 6,34 | 57.44 ± 7.12 | 0.715 |
| Gender (female) | 72 (68.6 %) | 70 (66.7 %) | 0.883 |
| Ethnicity (European) | 104 (99.0 %) | 105 (100 %) | 1.000 |
| Bodyweight, kg | 85.67 ± 12.92 | 86.94 ± 10.95 | 0.539 |
| BMI, kg/m2 | 31.17 ± 3.27 | 30.97 ± 2.74 | 0.213 |
| Smokers • Yes • No • Prior | 6 (5.7 %) 94 (89.5 %) 5 (4.8 %) | 10 (9.5 %) 89 (84.8 %) 6 (5.7 %) | 0.541 |
| Duration of diabetes, years | 10.24 ± 6.29 | 10.03 ± 6.45 | 0.731 |
| Oral antidiabetic drugs | 92 (87.6 %) | 96 (91.4 %) | 0.368 |
| Sulphonylurea treatment • No • Yes, prior the randomization • Yes, continued after the randomization | 78 (74.3) 21 (20.0) 6 (5.7) | 62 (59.1) 33 (31.4) 10 (9.5) | 0.064 |
| Prior insulin use • Yes • No | 74 (70.5%) 31 (29.5%) | 78 (74.3%) 27 (25.7%) | 0.643 |
| Prior insulin therapy: • Basal • Premixed insulins • Basal/bolus • Only bolus | (n = 74) 32 (43.2 %) 21 (28.4 %) 18 (24.3 %) 3 (4.1 %) | (n = 78) 50 (64.1 %) 15 (19.2 %) 13 (16.7 %) 0 (0.0 %) | 0.032 |
| HbA1c, % | 9.37 ± 1.01 | 9.51 ± 1.13 | 0.538 |
| FPG, mmol/l | 11.89 ± 3.70 | 11.81 ± 3.56 | 0.710 |
| Total insulin dose, units/day | 40.07 ± 20.72 | 35.35 ± 19.85 | 0.769 |
| AIA concentration, units/ml (at screening) | 2.74 ± 1.48 | 2.74 ± 1.26 | 0.459 |
| DTSQs, total score (at randomization) | 20.76 ± 8.06 | 21.02 ± 7.73 | 0.719 |
DTSQs: Diabetes Treatment Satisfaction Questionnaire – status; FPG: Fasting plasma glucose.
Immunogenicity
Frequency of immune response at week 26 did not differ between the groups (Table 2).
| Immunogenicity outcomes | GP-Lis25 (n = 105) | Ly-Lis25 (n = 105) | p-value |
|---|---|---|---|
| Patients (n) meeting the criteria of immune response • At week 12 • At week 26 | 14 (13.33 %) 9 (8.57 %) | 11 (10.48 %) 4 (3.81 %) | 0.651 0.164 |
| Change of AIA concentration from baseline, units/ml • At week 12 • At week 26 | 3.38 ± 13.05 (p = 0.427) 2.73 ± 11.46 (p = 0.056) | 1.05 ± 5.99 (p = 0.016) 0.63 ± 4.86 (p = 0.219) | 0.075 0.064 |
Mean AIA concentration at week 12 was 6.13 ± 12.93 units/ml in GP-Lis25 group and 3.78 ± 6.02 units/ml in Ly-Lis25 group. By week 26, mean AIA concentration was 5.48 ± 11.24 units/ml in GP-Lis25 group and 3.37 ± 4.82 units/ml in Ly-Lis25 group. Changes in AIA levels by weeks 12 and 26 are shown in Table 2. These changes were not statistically significant in both groups.
Neither AIA concentration at week 26, nor change in AIA concentration by week 26 from baseline influenced treatment efficacy (p = 0.734 and p = 0.727, respectively) and insulin dose (p = 0.282 for AIA concentration at week 26 and p = 0.378 for change in AIA concentration by week 26).
Blood samples of 12 patients in GP-Lis25 group and eight patients in Ly-Lis25 group having more than fourfold increase of AIA concentration at week 12, or at week 26, or both, were tested for insulin neutralizing antibodies. Three out of 12 patients in GP-Lis25 group and three out of eight patients in Ly-Lis25 group developed neutralizing AIAs during the study period, but none of them had any clinical signs showing that efficacy of insulin treatment was reduced. GP-Lis25 and Ly-Lis25 groups did not differ significantly in incidence of neutralizing AIA formation (p = 0.642).
Efficacy
HbA1c level was significantly decreased from baseline at week 12 for 1.36 ± 1.22% in GP-Lis25 group (p < 0.001) and for 1.34 ± 1.16% in Ly-Lis25 group (p < 0.001). HbA1c level was significantly decreased from baseline at week 26 for 1.59 ± 1.23% in GP-Lis25 group (p < 0.001) and for 1.60 ± 1.21% in Ly-Lis25 group (p < 0.001). These changes did not differ between the groups either at week 12 (p = 0.192), or at week 26 (p = 0.381). HbA1c changes are shown in Figure 2.
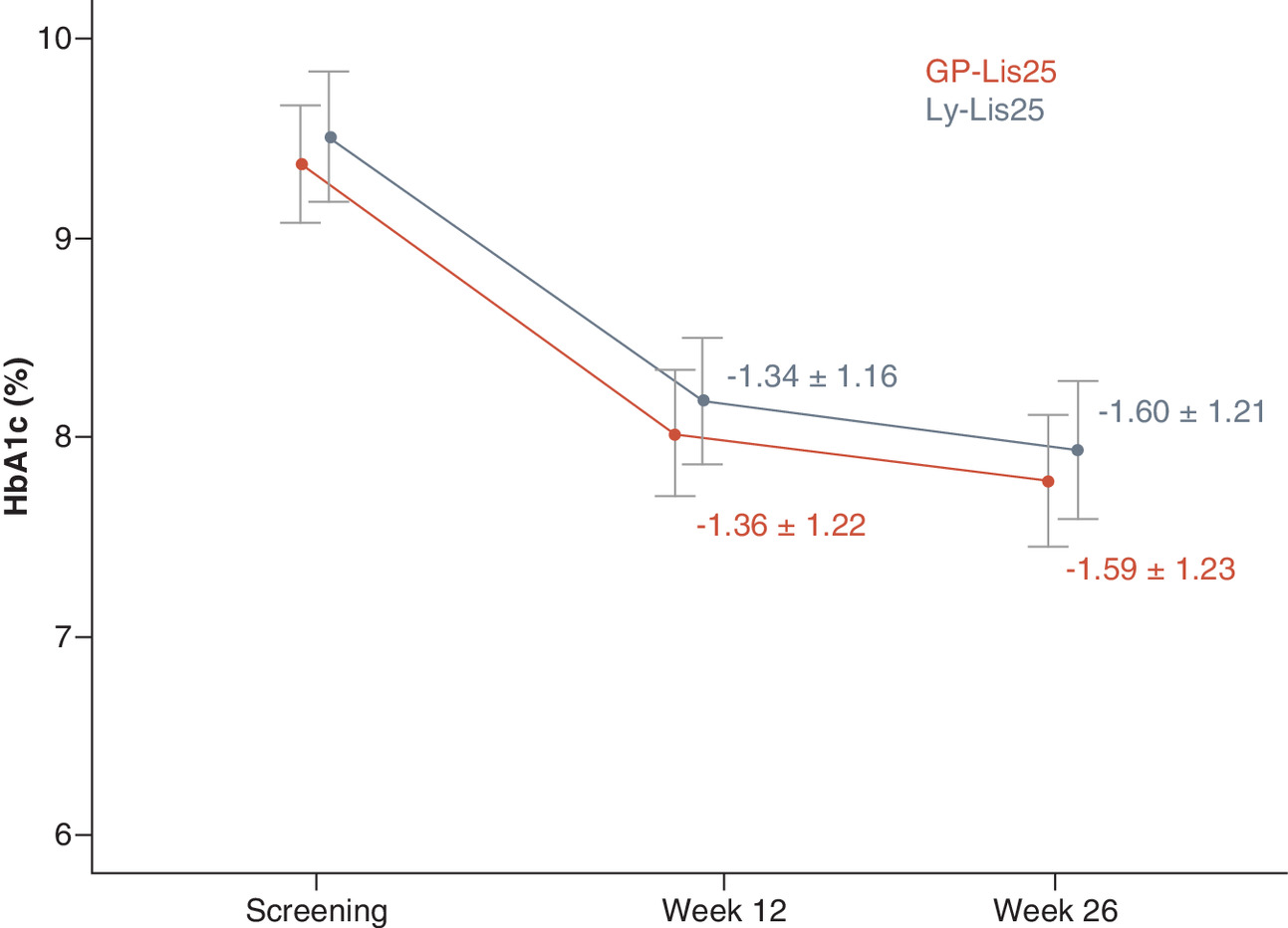
Intergroup difference of HbA1c level change at week 26 from baseline was (95% CI) 0.01 (-0.27–0.28)%. The CI does not cross the established 0.4% noninferiority margin demonstrating that GP-Lis25 is noninferior to Ly-Lis25 in terms of hypoglycemic efficacy.
Twenty-six patients (25%) in GP-Lis25 group and 23 (22%) patients in Ly-Lis25 group could achieve the pre-established individual glycemic goal at week 26 (p = 0.714).
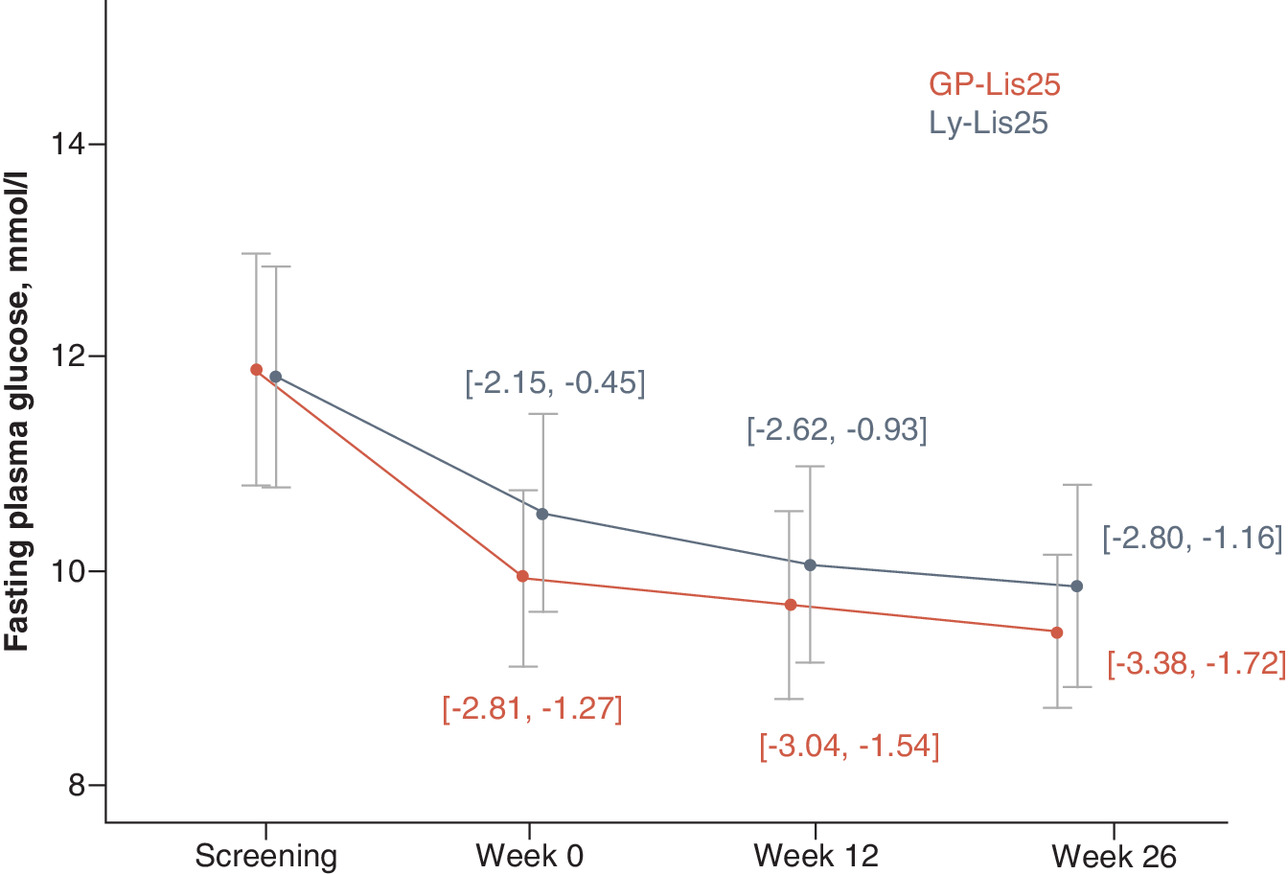
FPG level was significantly decreased from baseline at week 12 for 2.29 ± 3.82 mmol/l in GP-Lis25 group (p < 0.001) and for 1.78 ± 4.35 mmol/l in Ly-Lis25 group (p = 0.001). FPG level was also decreased significantly from baseline at week 26 for 2.55 ± 4.24 mmol/l in GP-Lis25 group (p = 0.001) and for 1.98 ± 4.22 mmol/l in Ly-Lis25 group (p = 0.001). These changes did not differ between the groups either at week 12 (p = 0.296), or at week 26 (p = 0.092). FPG changes are shown in Figure 3.
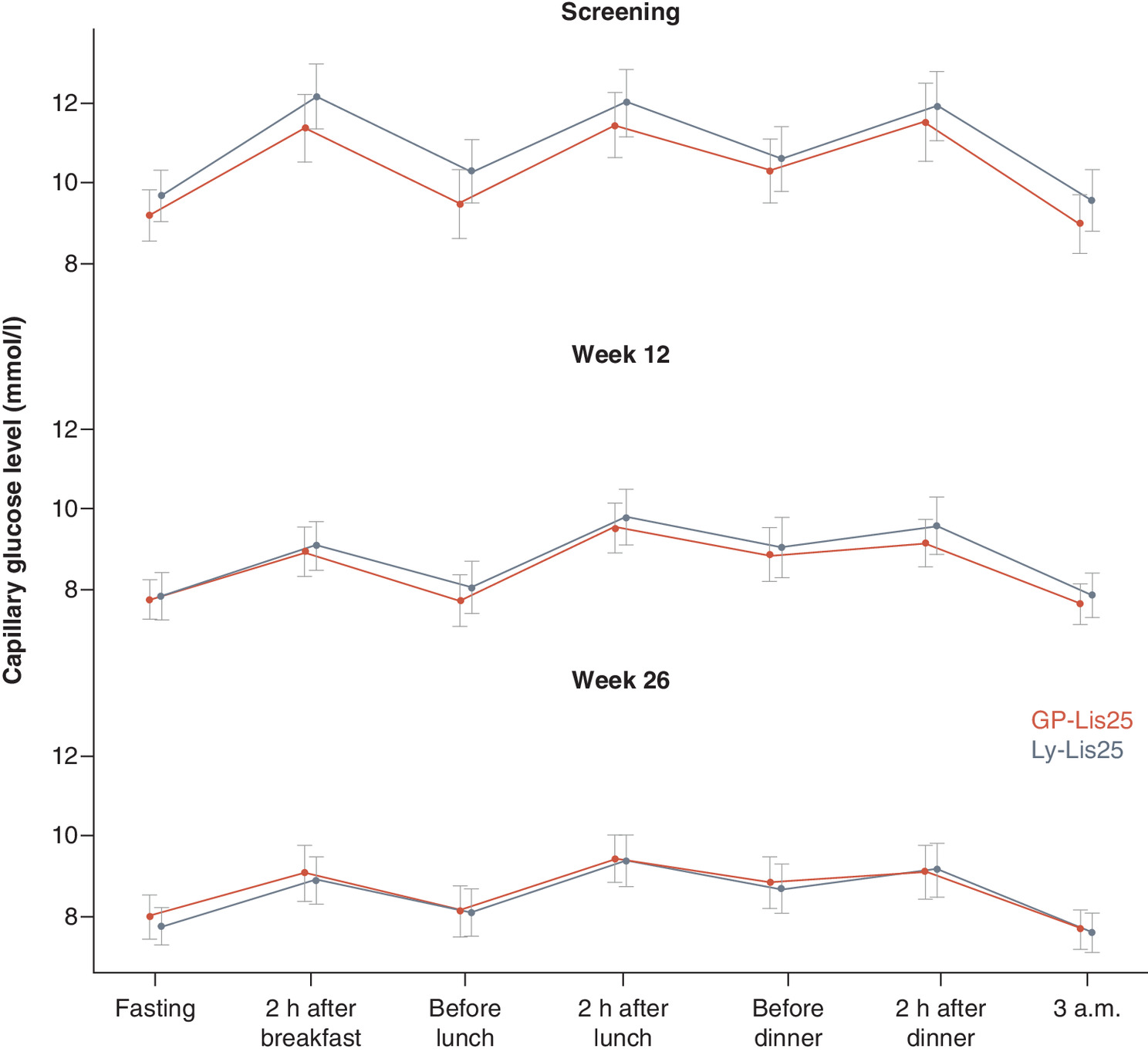
SPGP changes did not differ between the groups either at week 12 (p = 0.896, p = 0.811, p = 0.208, p = 0.432, p = 0.569, p = 0.214, p = 0.423 in fasting condition, two hours after breakfast, before lunch, two hours after lunch, before dinner, two hours after dinner and 3 a.m., respectively), or at week 26 (p = 0.407, p = 0.492, p = 0.832, p = 0.639, p = 0.801, p = 0.748, p = 0.762, for the abovementioned points, respectively). SPGP changes are shown in Figure 4.
Total insulin dose was significantly increased in both groups at week 26 compared with week 4 for 1.70 ± 5.97 units/day in GP-Lis25 group (p = 0.004) and 1.36 ± 4.79 units/day in Ly-Lis25 group (p = 0.005), but these changes did not differ between the groups (p = 0.878). The changes in basal insulin dose and bolus insulin dose were also not statistically significant and did not differ between the groups (p = 0.878 and p = 0.892, respectively).
Body weight of the patients was increased at week 26 compared with baseline values for 0.69 ± 2.33 kg in GP-Lis25 group (p = 0.004) and for 0.63 ± 2.29 kg in Ly-Lis25 group (p = 0.004). These changes did not differ between the groups (p = 0.832).
Treatment satisfaction was increased in both groups at week 26. The increase in DTSQs total score was 7.72 ± 8.44 points in GP-Lis25 group (p < 0.001) and 7.89 ± 8.65 points in Ly-Lis25 group (p < 0.001) and was similar between the groups (p = 0.258). DTSQc scores were 11.02 ± 6.68 points in GP-Lis25 group and 12.05 ± 6.28 points in Ly-Lis25 group and did not differ between the groups (p = 0.308).
Safety
The incidence of AEs, SAEs, discontinuations due to AEs, and adverse drug reactions reported in GP-Lis25 group were similar to those in Ly-Lis25 group (Table 3). One patient in GP-Lis25 group discontinued the clinical trial because of an AE (stroke, not related to the study treatment, the patient met the exclusion criteria). The relationship of adverse drug reactions to the investigational drug was assessed by the investigators as unlikely in all 6 cases (5 in GP-Lis25 group and 1 in Ly-Lis25 group).
| GP-Lis25 (n = 105) | Ly-Lis25 (n = 105) | |
|---|---|---|
| Patients with one or more AEs (n) | 38 (36.2 %) | 32 (30.5 %) |
| AEs (n) (total) • Mild • Moderate • Severe | 57 44 12 1 | 48 38 10 0 |
| Adverse drug reactions (n) | 5 | 1 |
| Serious AEs (n) | 4 | 1 |
| AEs of special interest (n) • Injection site reactions • Hypersensitivity reactions | 1 0 | 0 0 |
AE: Adverse event.
Most of these AEs were mild. There were five SAEs: four in GP-Lis25 group and one in Ly-Lis25 group, none of them were related to the study drugs. Three out five study participants with SAEs continued the study therapy, whereas two of them stopped using the investigational insulin drug.
One patient had one injection site reaction in GP-Lis25 group, and patients in Ly-Lis25 group did not develop injection site reactions. No hypersensitivity reactions were observed in either of the groups. There were no differences in any laboratory safety values or vital signs between the groups (data not shown).
Table 4 contains the summary of hypoglycemic episodes. The number of patients experiencing hypoglycemic episodes did not differ between the groups (p = 0.782). Ly-Lis25 group had less hypoglycemic episodes for 2.32 events/patient year. There were no severe hypoglycemic episodes (loss of consciousness, need in other people assistance) during the study. The investigators also evaluated severity of the episodes depending on a clinical situation, regardless the need in assistance of other people (in the same way they evaluated severity of any AEs). Most patients in both groups experienced mild hypoglycemia. Most hypoglycemic episodes were symptomatic, and there were less asymptomatic episodes in GP-Lis25 group (p < 0.001), but the number of patients having symptomatic or asymptomatic episodes did not differ between the groups (p = 0.344). There were no major (required external help) hypoglycemic episodes in this trial.
| Characteristics of hypoglycemic episodes | GP-Lis25 (n = 105) | Ly-Lis25 (n = 105) | |||
|---|---|---|---|---|---|
| Subjects (n) (%) | Events (n) (incidence rate/patient-year) | Subjects (n) (%) | Events (n) (incidence rate/patient-year) | ||
| Total patient-years | 50.75 | 51.07 | |||
| Total (<3.9 mmol/l) <3.0 mmol/l | 54 (51.4) 24 (22.9) | 452 (8.91) 97 (1.91) | 57 (54.3) 21 (20.0) | 316 (6.19) 39 (0.76) | |
| Severity (as of physician estimation) | |||||
| Mild | 52 (49.5) | 426 (8.39) | 55 (52.4) | 310 (6.07) | |
| Moderate | 4 (3.8) | 11 (0.22) | 2 (1.9) | 2 (0.04) | |
| Severe | 0 (0.0) | 0 (0) | 0 (0.0) | 0 (0) | |
| Symptoms | |||||
| Present | 49 (46.7) | 358 (7.05) | 43 (41.0) | 194 (3.75) | |
| Unknown | 3 (2.9) | 7 (0.14) | 1 (1.0) | 1 (0.02) | |
| Absent | 21 (20.0) | 87 (1.71) | 27 (25.7) | 121 (2.34) | |
| Time of the episode | |||||
| Night (00:00–05:59) | 23 (21.9) | 49 (0.97) | 8 (7.6) | 24 (0.46) | |
| Day (06:00–23:59) | 53 (50.5) | 403 (7.94) | 57 (54.3) | 292 (5.64) | |
Discussion & recommendations
The study was conducted to establish similarity of GP-Lis25 and Ly-Lis25 in terms of immunogenicity and safety and to confirm similar efficacy of the drugs.
Similar immunogenicity was confirmed based on AIA and partial neutralizing AIA assessment using all pre-established immunogenicity end points. We did not observe any AIA formation that might lead to any change in efficacy of insulin lispro biphasic 25 or reduce safety of the drug. Both GP-Lis25 and Ly-Lis25 were generally well tolerated, and there was no evidence of clinically significant differences with regards to their safety profiles. AIA did not influence either treatment efficacy, or insulin dose.
Similar safety was confirmed based on AEs assessment, as no significant differences in AEs and SAEs between GP-Lis25 and Ly-Lis25 groups were recorded. There were less hypoglycemic episodes in Ly-Lis25 group, however, absolute difference in events per patient-year (2.32 events/patient-year) was comparable with the incidence rates observed in other studies of insulin biosimilars in T2D [12,13]. During blinded medical monitoring of the study some patients with inadequate insulin doses were revealed: they reported more hypoglycemic episodes than the most of other study subjects, and they had low glycemic values during SPGP. The medical monitor of the study pointed this out for the responsible investigators; however, the Sponsor did not influence investigators’ decisions. At the point of data analysis, there were more patients with frequent hypoglycemic episodes in GP-Lis25 group.
Similar efficacy was confirmed based on HbA1c level at weeks 12 and 26, and it was supported by other efficacy end points, such as FPG and SPGP. FPG changes from baseline at week 12 and 26 were significant in both groups, and they did not differ between GP-Lis25 and Ly-Lis25 groups. SPGP changes did not differ between groups either at week 12, or at week 26.
GP-Lis25 and Ly-Lis25 doses were not changed significantly during the treatment period, and these changes did not differ between the groups. Patients’ body weight was changed similarly without any statistically significant difference in both treatment groups. The results of DTSQs and DTSQc questionnaires confirmed similar treatment satisfaction with GP-Lis25 and Ly-Lis25 treatment.
Thus, this study demonstrated a high degree of clinical similarity of the study drugs.
Taking into account all the data received from the head-to-head comparative preclinical studies (physical and chemical properties, in vitro pharmacodynamics), comparative euglycemic clamp study [7] and the results of this comparative safety (immunogenicity) trial, GP-Lis25 biosimilarity to Ly-Lis25 was proved in accordance with the regulatory guidelines.
Since the trial data can be extrapolated to GP-Lis as a less immunogenic insulin preparation, and all other evidence prove its similarity to the reference drug Humalog, GP-Lis has also proved its biosimilarity to Humalog.
Future perspective
Since original biologics requires an extensive investigational program, these drugs are usually rather expensive, and many patients cannot afford them without insurance programs. Biosimilar products require less clinical trials because efficacy and safety of active pharmaceutical substance has already been proved by originator product, and the main goal of all biosimilar investigational program is to show that biosimilar does not differ from original biological. A cheaper investigational program leads to possibility of establishing a lower price for biosimilar products as compared with original biologicals, and it can be crucial in some countries with expensive biological drugs. Biosimilars is the opportunity for more and more people to get access to high-quality effective drugs for their better health and better future.
Study limitations
1.
We included only T2D patients, since the use of premixed insulins is not recommended in T1D population [1]. However, the data on immunogenicity and safety can be rightfully extrapolated to patients with T1D.
2.
This study was open-label, and it could influence on investigator approach to insulin dose titration. However, HbA1c level was taken as a secondary end point since it is not sensitive enough to establish similar efficacy in comparison with glucose infusion rate, estimated in euglycemic clamp studies. Blinding of these studies is impossible due to specific signs on insulin delivery systems and even not required because immunogenicity end points are objective, and open-label design cannot influence it [4,5].
3.
4.
Only white Caucasians were included into the study. However, according to pharmacology principles, if two medicines show no difference in one subpopulation, they will show no difference in any other population (based on this principle, bioequivalence studies on healthy males are usually enough for generic drug authorization). So, there is no point of concern regarding limitations of the included ethnicity.
•
Clinical development program of biosimilars is strictly regulated.
•
Safety (immunogenicity) and efficacy study is the last stage of a biosimilar clinical development program.
•
If a biosimilar manufacturer develops different preparations containing the same active ingredient, the drug with the highest expected immunogenic potential should be included in the safety study.
•
The primary objective of the current study was to compare immunogenicity of GP-Lis25 (biosimilar) and Ly-Lis25 (original biological).
•
A pivotal efficacy study for biosimilar insulins is euglycemic clamp study, whereas the current study is confirmatory in terms of efficacy.
•
GP-Lis25 was similar to Ly-Lis25 in terms of immunogenicity, safety and efficacy based on the established end point.
•
The data can be extrapolated to GP-Lis in accordance with regulatory guideline.
Acknowledgments
The authors thanked AS Agafina, MB Antsiferov, OK Khmelnitsky, ND Krasnopeeva, TI Kulagina, OE Lantseva, ZV Paltsman, VV Popov, TI Rodionova, JG Samoilova, AF Verbovoy, SV Vorobiev, KA Zakharov, OV Zanozina, NV Zhavoronkova and all investigational teams for taking part in this study. The authors also thanked Unimed Laboratories and Exacte Labs for laboratory testing.
Financial & competing interests disclosure
This study was funded by OOO GEROPHARM, Russia. AA Mosikian, IE Makarenko, IS Lunev, BR Zinnatulina and RV Drai are current employees of OOO GEROPHARM, Russia. AY Mayorov, DN Alpenidze and VL Orlova were principal investigators, and MV Verbovaya was a co-investigator in this study. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Data sharing statement
The authors certify that this manuscript reports original clinical trial data (NCT04023344). The data will not be made publicly available unless it is personally requested from the principal investigator.
Supplementary Material
File (suppl_file.docx)
- Download
- 13.97 KB
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
1.
Dedov II, Shestakova MV, Mayorov AY et al. Standards of specialized diabetes care (9th edition). Diabetes Mellitus 22(1S1), 1–144 (2019).
• All the medicinal features of the clinical trial protocol are based on this document.
2.
EMA. Biosimilar medicines: overview (EMA) (2020). www.ema.europa.eu/en/human-regulatory/overview/biosimilar-medicines-overview
3.
Mulcahy AW, Hlavka JP, Case SR. Biosimilar cost savings in the United States: initial experience and future potential. Rand Health Q. 7(4), 3 (2018).
4.
Resolution No. 89 of November 3, 2016 On Approval of the Rules on Conduct of Studies of Biological medicines in the Eurasian Economic Union, Chapter 15.7 Preclinical and clinical development of bioanalogue (biosimilar) medicinal products containing recombinant insulin and insulin analogues. (2016).
•• Main regulatory guidance for biosimilar insulin registration in the Eurasian Economic Union.
5.
European Medical Agency. Guideline on non-clinical and clinical development of similar biological medicinal products containing recombinant human insulin and insulin analogues. EMEA/CHMP/BMWP/32775/2005_Rev. 1 (2015). www.ema.europa.eu/en/documents/scientific-guideline/first-draft-guideline-non-clinical-clinical-development-similar-biological-medicinal-products_en.pdf
• Original regulatory guidance for registration of insulin biosimilars in the European Union.
6.
Mayorov A Yu, Fedotov IA, Drai RV, Avdeeva OI, Makarenko IE. Results of the Estimation of Biosimilarity of RinLiz® (LLC «GEROPHARM», Russia) and Humalog® (Lilly France, France) Using the Method of the Hyperinsulinemic Eulygemic Clamp on Healthy Voluntary. Drug development & registration. 9(2), 124–131 (2020).
7.
Mayorov AY, Koksharova EO, Mishina EE et al. Assessment the equivalence of the bioanalogue insulin lizpro biphasic 25 (Geropharm-bio, Russia) and Humalog Mix 25 (Lilly France, France) using the euglycemic hyperinsulinum clamp method on healthy volonters. Diabetes Mellitus 21(6), 462–471 (2018).
8.
Abramenko NB, Vnukova PI, Golovina ES et al. Development and validation of approach for the detection of neutralizing antibodies against insulin (glargine) in human blood plasma. Drug Devel. Registration 8(3), 70–78 (2019).
9.
Dedov II, Shestakova MV, Mayorov AY et al. Standards of specialized diabetes care (8th Edition). Diabetes Mellitus 20(S1), 1–121 (2017).
10.
European Medical Agency. Guideline on clinical investigation of medicinal products in the treatment or prevention of diabetes mellitus. CPMP/EWP/1080/00 Rev. 1 (2012). www.ema.europa.eu/en/documents/scientific-guideline/guideline-clinical-investigation-medicinal-products-treatment-prevention-diabetes-mellitus-revision_en.pdf
11.
World Medical Association. World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA 310, 2191–2194 (2013).
12.
Derwahl KM, Bailey TS, Wernicke-Panten K et al. Efficacy and safety of biosimilar SAR342434 insulin lispro in adults with Type 2 diabetes, also using insulin glargine: SORELLA 2 study. Diabetes Technol. Ther. 20(1), 49–58 (2018).
13.
Hollander PA, Carofano WL, Lam RLH et al. Efficacy and safety of MK-1293 insulin glargine compared with originator insulin glargine (Lantus) in Type 2 diabetes: a randomized, open-label clinical trial. Diabetes Obes. Metab. 20(9), 2229–2237 (2018).
Information & Authors
Information
Published In
Pages: 55 - 66
PubMed: 33355484
Copyright
© 2020 Future Medicine Ltd.
History
Received: 25 April 2020
Accepted: 27 October 2020
Published online: 23 December 2020
Keywords:
Topics
Authors
Metrics & Citations
Metrics
Article Usage
Article usage data only available from February 2023. Historical article usage data, showing the number of article downloads, is available upon request.
Citations
How to Cite
Efficacy and safety of GP40021 insulin lispro biphasic compared with Humalog Mix 25 in Type 2 diabetes mellitus patients. (2020) Journal of Comparative Effectiveness Research. DOI: 10.2217/cer-2020-0064
Export citation
Select the citation format you wish to export for this article or chapter.
Citing Literature
- Xiaoxuan Xing, Lingyi Zhao, Ke Wang, Zhizhou Wang, Lan Zhang, Xianzhe Dong, Therapeutic equivalence and switching between biosimilar and reference insulins: A systematic review and meta‐analysis of randomised controlled trials, Diabetes, Obesity and Metabolism, 10.1111/dom.70328, 28, 2, (1371-1382), (2025).
- N. A. Petunina, I. A. Kuzina, M. E. Telnova, E. V. Goncharova, N. S. Martirosyan, A. O. Shchetinina, M. V. Khachaturov, Insulin therapy for elderly patients with type 2 diabetes mellitus, Meditsinskiy sovet = Medical Council, 10.21518/ms2024-127, 6, (16-22), (2024).
- Ekaterina Koksharova, Roman Drai, Sergei Noskov, Artem Dorotenko, Ekaterina Protsenko, Kseniia Radaeva, Anna Arefeva, Maria Gefen, Gagik Galstyan, Igor Makarenko, Clinical Pharmacology of GP40321 (Insulin Glulisine Biosimilar): Pharmacokinetic and Pharmacodynamic Comparability in a Hyperinsulinemic‐Euglycemic Clamp Procedure, Clinical Pharmacology in Drug Development, 10.1002/cpdd.1401, 13, 7, (828-836), (2024).
- Thomas Danne, Lutz Heinemann, Jan Bolinder, New Insulins, Biosimilars, and Insulin Therapy, Diabetes Technology & Therapeutics, 10.1089/dia.2022.2503, 24, S1, (S-35-S-57), (2022).
- Li-Jou Yang, Ta-Wei Wu, Chao-Hsiun Tang, Tzu-Rong Peng, Efficacy and immunogenicity of insulin biosimilar compared to their reference products: a systematic review and meta-analysis, BMC Endocrine Disorders, 10.1186/s12902-022-00944-5, 22, 1, (2022).