Ataluren use in patients with nonsense mutation Duchenne muscular dystrophy: patient demographics and characteristics from the STRIDE Registry
Publication: Journal of Comparative Effectiveness Research
Abstract
Aim: Strategic Targeting of Registries and International Database of Excellence (STRIDE) is an ongoing, multicenter registry providing real-world evidence regarding ataluren use in patients with nonsense mutation Duchenne muscular dystrophy (DMD) in clinical practice (NCT02369731). Here, we describe the initial demographic characteristics of the registry population. Patients & methods: Patients will be followed up from enrollment for ≥5 years or until study withdrawal. Results & conclusion: As of 9 July 2018, 213 DMD boys were enrolled from 11 countries. Mean (standard deviation) ages at first symptoms and at study treatment start were 2.7 (1.7) years and 9.8 (3.7) years, respectively. Corticosteroids were used by 190 patients (89.2%) before data cut-off. Mean (standard deviation) ataluren exposure was 639.0 (362.9) days. Six patients withdrew. STRIDE is the first drug registry for patients with DMD and represents the largest real-world registry of patients with nmDMD to date.
Duchenne muscular dystrophy (DMD) is a rare, progressive, X-linked neuromuscular disorder that occurs in approximately one in every 3600–5000 live male births [1–3]. It is characterized by progressive muscle degeneration and weakness due to the absence of functional dystrophin protein [3,4]. Patients with DMD experience functional decline, loss of ambulation, wheelchair dependency, and ultimately early death due to respiratory or cardiac failure [2,3]. In approximately 10–15% of boys with DMD, the disorder is caused by a nonsense mutation in the DMD gene [4–6]. This mutation creates a premature stop codon in the dystrophin mRNA, which prevents the production of full-length, functional dystrophin protein [7]. The presence of a nonsense mutation in the DMD gene should be determined by genetic testing [8].
Ataluren is a first-in-class, orally bioavailable treatment for patients with nonsense mutation DMD (nmDMD), designed to promote ribosomal readthrough of an in-frame premature stop codon and thereby enable the production of full-length dystrophin protein [9,10]. Ataluren has been evaluated in patients with nmDMD in two randomized, double-blind, placebo-controlled trials: one Phase Ib trial [11] and one Phase III trial (Ataluren Confirmatory Trial in DMD [ACT DMD]) [12]. Both trials showed that ataluren (40 mg/kg/day) had favorable efficacy compared with placebo in a range of functional end points over 48 weeks, despite not meeting the primary end points [11,12]. In these trials, ataluren was generally well tolerated and the majority of treatment-emergent adverse events (AEs) were mild to moderate in severity, indicating that ataluren has a favorable benefit–risk profile over a 48-week study period.
Ataluren is indicated for the treatment of nmDMD in ambulatory patients aged 5 years or older in Brazil, Chile, Israel, the Republic of Korea and Ukraine, and more recently the indication has expanded to include patients aged 2 years or older in member states of the European Union and Iceland, Liechtenstein and Norway [8]. Efficacy has not yet been demonstrated in nonambulatory patients [8].
To fulfill a postmarketing commitment to the Pharmacovigilance Risk Assessment Committee of the European Medicines Agency, and in an effort to generate data on patterns of use and long-term patient outcomes in the setting of real-world routine clinical practice, an observational registry assessing the safety and effectiveness of ataluren is currently under way. This registry, the Strategic Targeting of Registries and International Database of Excellence (STRIDE; ClinicalTrials.gov identifier: NCT02369731), constitutes the first drug registry for patients with DMD. Here, we describe the initial demographic characteristics of the study population in the STRIDE Registry, as of 9 July 2018.
Patients & methods
Study design
The STRIDE Registry is an ongoing, multicenter, observational, postapproval safety study of ataluren in patients with nmDMD (ClinicalTrials.gov identifier: NCT02369731). The study is being conducted in countries where ataluren is available commercially or through an early access program; additional countries may be added as ataluren becomes more widely accessible. To fulfill regulatory requirements, enrolled patients will be followed up for at least 5 years from the date of enrollment or until withdrawal from the study or death, whichever occurs first [13]. Patients, who discontinue treatment with ataluren, will continue to be followed up for the duration of the study, unless they withdraw consent to participate.
No study medication is provided as part of this observational registry study; the treating physician is responsible for all treatment decisions according to their usual practice and provides prescriptions as appropriate. There are no standard protocol-mandated procedures or mandated diagnostic tests, although physicians and caregivers are encouraged to follow published treatment guidelines and standards of care, and nonsense mutations must be identified by genetic testing at an accredited laboratory [3,8,14,15]. Patients, who do not have a nonsense mutation in the DMD gene, should not receive ataluren [8].
Ethics
Before any patient is enrolled and study-related data are collected, ethics committee approval of the protocol, written informed consent form and all patient enrollment materials are obtained in each country and for each site, as applicable. When appropriate, assent was obtained from the patient. The study is conducted in accordance with the ethical principles outlined in the Declaration of Helsinki, applicable privacy laws and local regulations for each participating site, as well as with the Guidelines for Good Pharmacoepidemiology Practices.
Patient eligibility
Patients with a confirmed genetic diagnosis of DMD are eligible to participate in the study if they are, or will be, receiving usual-care treatment with a commercial supply of ataluren (or receiving care within an early access program), and are willing to provide written informed consent (by themselves or their parent/legal guardian) for data-collection procedures.
Patients are excluded from the study if they are receiving ataluren or placebo in an ongoing blinded, randomized clinical trial, or if they are receiving ataluren in any other ongoing clinical trial or early access program that prevents participation in the current study. Patients receiving ataluren in a clinical trial or in an early access program can become eligible for this study once they have completed the required follow-up and fulfilled the conditions of the trial or program. Note, patients who are nonambulatory at study entry and are receiving a commercial supply of ataluren had previously received ataluren in a clinical trial during which they were ambulatory.
Study data
Effectiveness information related to ataluren use (40 mg/kg/day; 10, 10 and 20 mg/kg for morning, mid-day and evening doses, respectively) in routine clinical care is documented and includes routine clinical assessments of motor, cardiac and pulmonary function (e.g., timed function tests, North Star Ambulatory Assessment [16], ambulation as assessed by the 6-min walk test, time to loss of ambulation, frequency of wheelchair use, or use of other ambulatory-based assistive devices, Performance of Upper Limb scale [17], and cardiac and pulmonary function assessments). Assessments are performed as per usual clinical care at each center and are not mandatory. This study is part of the risk-management plan for ataluren; therefore, safety information collected during routine clinical practice, including AEs, changes in laboratory tests of interest, and information regarding special events of interest (as per the risk-management plan), is also collected. The safety of ataluren in patients enrolled in the STRIDE Registry, and effectiveness results for propensity-score matched populations from STRIDE and the Cooperative International Neuromuscular Research Group Duchenne Natural History Study (CINRG DNHS), will be presented in a separate publication. Effectiveness data from the STRIDE Registry will be compared with those from the CINRG DNHS using Kaplan–Meier estimates and Cox regression for motor, pulmonary and cardiac function outcomes.
Data collection & study methodology
A summary of the STRIDE Registry study design and methodology is shown in Figure 1. At the inclusion visit, once patients provide their or their parent’s/guardian’s informed consent to be enrolled in the registry, data are collected, including demographic characteristics, diagnosis information, medical history (e.g., ambulation status, previous and current exposure to ataluren, and prior and concomitant medications), and clinical information and symptoms. Loss of ambulation is defined as full-time wheelchair requirement, and prior medications are defined as any medications that patients ended before study entry. For patients who initiated ataluren as part of a commercial or early access program before enrollment in the registry, data are obtained retrospectively from their medical records for the time period between ataluren initiation and enrollment, with appropriate patient/parent/guardian consent (retrospective visits). Retrospective data from previous clinical trials will not be utilized. Patients enrolled in the study will continue to be followed up for at least 5 years from the date of enrollment or until withdrawal from the study, and data will be collected during routine follow-up visits conducted as per usual care, which are estimated to occur at 3- to 6-month intervals. At each follow-up visit, data will be collected on survival outcomes; status of ambulation; clinical information, including effectiveness and safety data; current ataluren treatment (e.g., dosing compliance); and concomitant medications, which are defined as any medications that patients started on or after the treatment start date. Assessments of musculoskeletal health, rehabilitation, orthopedic and gastrointestinal management, as well as other measures of psychosocial management, will be collected to allow for comparison of patient health-management activities in routine clinical care to those of published treatment guidelines. The data cut-off for inclusion in the present analyses was on 9 July 2018.
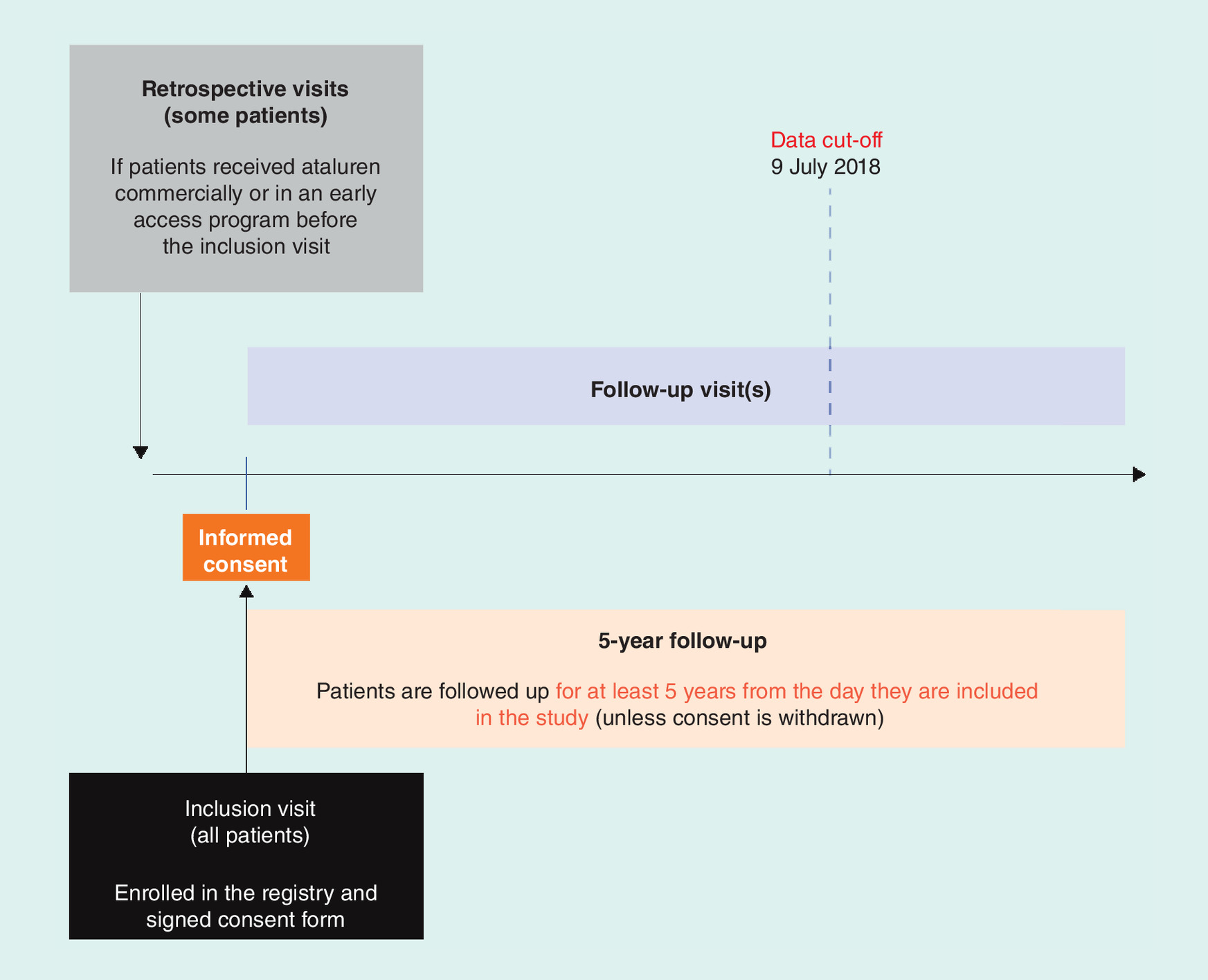
Study data are recorded in an electronic data capture system or in paper case-report forms by the treating physician or designee during a clinic visit or from hospital records, clinical and office charts, and evaluation checklists. All data are entered via secure web-based data-entry screens and transferred via Secure Sockets Layer connections to the study database.
Statistical analysis
Originally, the study was intended to enroll approximately 200 patients, and enrollment was to conclude upon entry of the 200th patient or the completion of the 2-year enrollment period, whichever occurred first. The enrollment target was chosen for practical and statistical reasons, taking into account the size of the population of patients with nmDMD in the European Union. The minimum exposure of patients who continue to receive ataluren and who do not withdraw from the study is 5 years; assuming that the rate of patients lost to follow-up is 2–5% per year, there would be a minimum of 1000 person-years of exposure to ataluren. The protocol was subsequently amended to enable enrollment of up to 270 patients because more patients were available for enrollment than expected.
Corticosteroid use was defined as the investigator checking ‘yes’ at any time during the study to the following case report form question: ‘is the participant currently taking or has ever taken any corticosteroid medication during the study protocol?’
In general, continuous variables were summarized using descriptive statistics, including the number of patients (n), mean, standard deviation (SD), 95% CIs, median, minimum and maximum. Categorical variables were summarized using frequency count as the number and percentage of patients.
Results
Patient disposition
Enrollment of the first patient in the STRIDE Registry occurred on 31 March 2015. As of 9 July 2018, 217 patients (213 male patients, three female patients and one patient who failed screening) with DMD (Figure 2) from 11 countries with 53 active study sites have been enrolled. The present report details demographic parameters among male patients who provided informed consent and met eligibility criteria (the evaluable population; n = 213). Female patients were excluded from this analysis because effectiveness data from patients in this population will be compared in a separate publication with those from an all-male cohort (CINRG DNHS). Demographics and characteristics of the female patients are included in Supplementary Table 1. Of the 213 patients in the evaluable population, nine patients with frameshift mutations were excluded from the effectiveness population because ataluren is not indicated in these patients) [8]. The number of active countries, number of study sites and distribution of patients per country are shown in Figure 3.
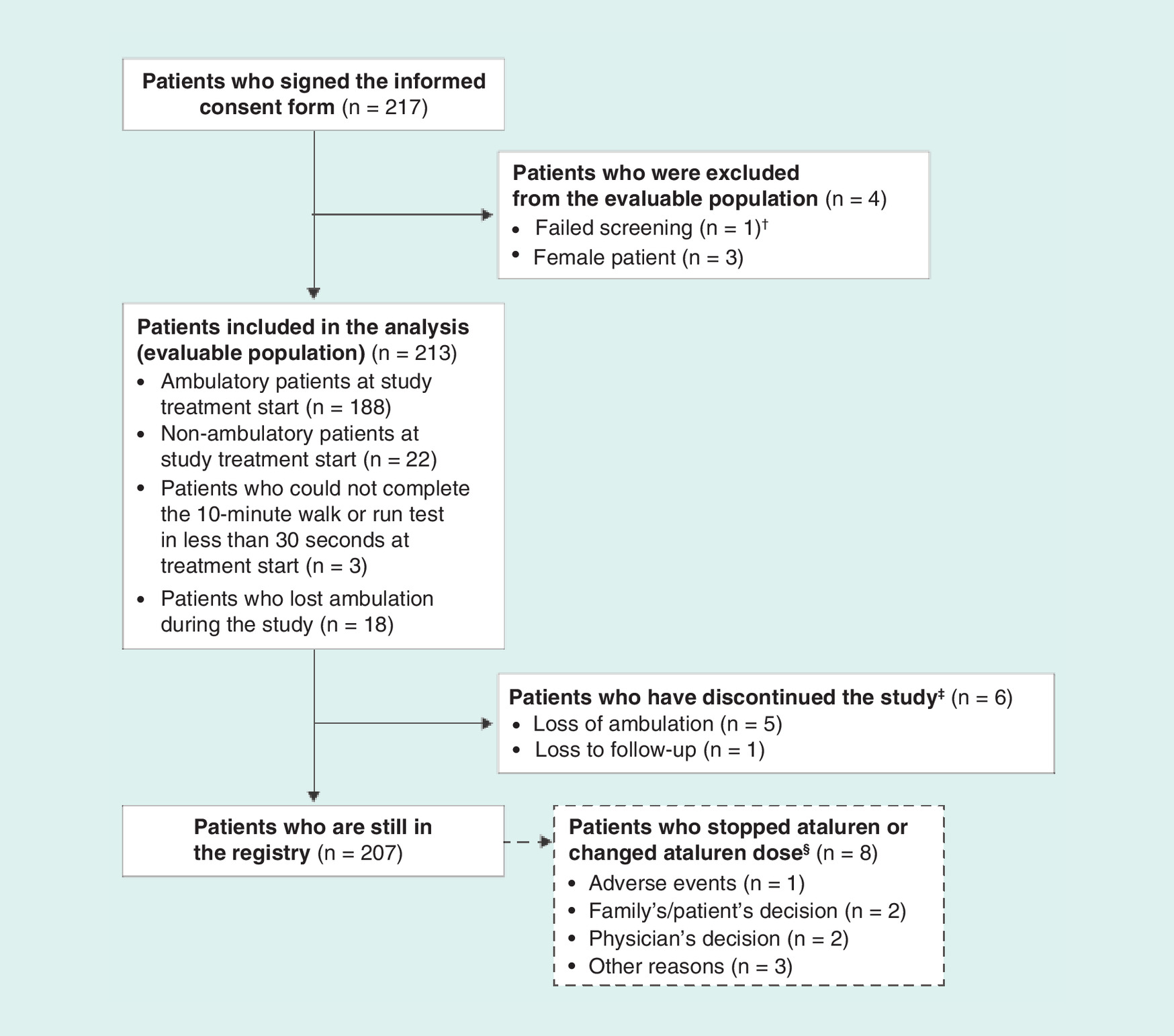
Figure 2. Patient disposition.
†Screening failure owing to a frameshift mutation.
‡Patients who withdrew from the study did not necessarily discontinue ataluren treatment.
§No patient changed ataluren dose.

A total of 61 out of 213 patients (28.6%) had participated in previous PTC Therapeutics clinical trials of ataluren before joining the registry (Table 1). A total of 190 patients (89.2%) received corticosteroids during the study and before data cut-off, whereas 23 patients (10.8%) had never received corticosteroids.
| Characteristic | Patients in the evaluable population (n = 213) |
|---|---|
| Sex, n (%) | |
| Male | 213 (100.0) |
| Race, n (%) | |
| White | 142 (66.7) |
| Arab/Middle Eastern | 6 (2.8) |
| Arab/Middle Eastern, Asian | 1 (0.5) |
| Asian | 5 (2.3) |
| Mixed race, black/white | 1 (0.5) |
| North African | 1 (0.5) |
| Latin | 1 (0.5) |
| Unknown | 54 (0.9) |
| Weight at first visit† captured within the registry (kg) | |
| n | 187 |
| Mean (SD) | 30.9 (13.4) |
| 95% CI | 29.0, 32.9 |
| Median | 26.5 |
| Min, max | 13.5, 87.0 |
| Height at first visit† captured within the registry (cm) | |
| n | 159 |
| Mean (SD) | 124.1 (16.1) |
| 95% CI | 121.6, 126.6 |
| Median | 121.1 |
| Min, max | 94.3, 178.0 |
| BMI at first visit† captured within the registry (kg/m2) | |
| n | 159 |
| Mean (SD) | 19.0 (4.3) |
| 95% CI | 18.4, 19.7 |
| Median | 17.9 |
| Min, max | 13.7, 40.5 |
| Age at first symptoms (years) | |
| n | 195 |
| Mean (SD) | 2.7 (1.7) |
| 95% CI | 2.4, 2.9 |
| Median | 2.5 |
| Min, max | 0.0‡, 8.0 |
| Age at muscle biopsy (years) | |
| n | 119 |
| Mean (SD) | 4.5 (2.5) |
| 95% CI | 4.1, 5.0 |
| Median | 4.2 |
| Min, max | 0.4, 13.0 |
| Time between first symptoms and muscle biopsy (years) | |
| n | 112 |
| Mean (SD) | 1.6 (1.9) |
| 95% CI | 1.3, 2.0 |
| Median | 1.1 |
| Min, max | -3.4, 8.0 |
| Age at genetic confirmation of nmDMD diagnosis (years) | |
| n | 202 |
| Mean (SD) | 5.2 (2.9) |
| 95% CI | 4.8, 5.6 |
| Median | 4.9 |
| Min, max | -0.4‡, 15.6 |
| Time between muscle biopsy and genetic confirmation of nmDMD diagnosis (years) | |
| n | 119 |
| Mean (SD) | 1.0 (2.2) |
| 95% CI | 0.6, 1.4 |
| Median | 0.3 |
| Min, max | -5.0, 12.3 |
| Time between first symptoms and/or laboratory abnormality and genetic confirmation of nmDMD diagnosis (years) | |
| n | 186 |
| Mean (SD) | 2.4 (2.4) |
| 95% CI | 2.0, 2.7 |
| Median | 1.7 |
| Min, max | -3.4, 12.6 |
| Previously enrolled in ataluren clinical trial, n (%) | |
| No | 152 (71.4) |
| Yes | 61 (28.6) |
| Age at informed consent (years) | |
| n | 213 |
| Mean (SD) | 10.5 (3.6) |
| 95% CI | 10.0, 11.0 |
| Median | 10.0 |
| Min, max | 5.0, 22.8 |
| Age category at informed consent (years), n (%) | |
| <5 | 0 (0.0) |
| ≥5 | 213 (100.0) |
| ≥5 to <7 | 37 (17.4) |
| ≥7 to <10 | 69 (32.4) |
| ≥10 to <15 | 81 (38.0) |
| ≥15 | 26 (12.2) |
| Age at first visit† captured within the registry (years) | |
| n | 213 |
| Mean (SD) | 9.8 (3.7) |
| 95% CI | 9.3, 10.3 |
| Median | 9.0 |
| Min, max | 4.0, 22.8 |
| Age category at first visit† captured within the registry (years), n (%) | |
| <5 | 3 (1.4) |
| ≥5 | 210 (98.6) |
| ≥5 to <7 | 55 (25.8) |
| ≥7 to <10 | 62 (29.1) |
| ≥10 to <15 | 72 (33.8) |
| ≥15 | 21 (9.9) |
| Age at cut-off date (years) | |
| n | 213 |
| Mean (SD) | 11.6 (3.6) |
| 95% CI | 11.1, 12.1 |
| Median | 11.5 |
| Min, max | 5.5, 24.2 |
†
First visit data are defined as the first data available for each patient, which can either be from a retrospective visit or from the inclusion visit when informed consent was obtained.
‡
A minimum age value of zero or a negative minimum age value is due to a prenatal diagnosis.
nmDMD: Nonsense mutation Duchenne muscular dystrophy; SD: Standard deviation.
At the patients’ treatment start date, otherwise known as the first visit captured within the registry, 188 patients (88.3%) were ambulatory, 22 patients (10.3%) were nonambulatory and three patients (1.4%) could not complete the 10-min walk or run test in less than 30 s. Of the 188 patients who were ambulatory at the first visit, 18 (9.6%) lost ambulation during the study.
Of the 213 enrolled boys, six have withdrawn from the study (n = 5, owing to loss of ambulation; n = 1, lost to follow-up), and eight have discontinued ataluren but remain in the registry (n = 1, AEs; n = 2, family’s/patient’s decision; n = 2, physician’s decision; n = 3, other reasons [e.g., the product could no longer be prescribed owing to local regulations]). Patients who withdrew from the study did not necessarily discontinue ataluren treatment; for example, the five patients who withdrew owing to loss of ambulation did not discontinue treatment.
Patient demographics & characteristics
The demographics and characteristics of patients in the study are shown in Table 1. Of the 213 boys in the registry, the majority were white (142 boys, 66.7%). The mean (SD) weight, height and BMI at informed consent (inclusion visit) were 30.9 (13.4) kg, 124.1 (16.1) cm and 19.0 (4.3) kg/m2, respectively.
The mean (SD) age at first symptoms, at the time of muscle biopsy and at genetic confirmation of nmDMD diagnosis was 2.7 (1.7) years, 4.5 (2.5) years and 5.2 (2.9) years, respectively. Of the 213 boys in the registry, 119 (55.9%) had a muscle biopsy. A list of genetically confirmed DMD mutations for patients in the STRIDE Registry is shown in Supplementary Table 2. There were nine patients whose age at first symptoms was 6 years or older, which is older than that typically expected for patients with DMD [2]; however, these patients had symptoms consistent with DMD, and seven of the patients had a stop codon mutation that has previously been identified and linked to a DMD phenotype (Supplementary Table 3). The mean (SD) times between first symptoms and muscle biopsy and between muscle biopsy and genetic confirmation of nmDMD diagnosis were 1.6 (1.9) years and 1.0 (2.2) years, respectively. The mean (SD) time between first symptoms and genetic nmDMD diagnosis was 2.4 (2.4) years.
The mean age (SD) of patients enrolled in the registry at the inclusion visit was 10.5 (3.6) years. At this time, the most common age category was ≥10 to <15 years (81/213 patients [38.0%]), and 150 patients (70.4%) were in the age category of ≥7 to <15 years. Patients first captured in the registry had a mean (SD) age of 9.8 (3.7) years. The mean (SD) age of patients at the cut-off date of 9 July 2018 was 11.6 (3.6) years.
The most common first clinical signs, symptoms and/or laboratory abnormalities experienced in the lifetime of the patients, which were reported at the inclusion visit, were elevated creatine kinase (54.0%), delayed walking (31.9%), difficulty going up stairs (31.0%), difficulty getting up from the floor (Gowers’ sign) (26.8%) and enlarged calves (pseudohypertrophy) (25.4%) (Supplementary Table 4).
Prior & concomitant medications
Total 48 (22.5%) patients received medications that ended before study entry, which included corticosteroids (13.1%), vitamin D and vitamin D analogs (3.8%), angiotensin-converting enzyme (ACE) inhibitors (2.3%) and calcium (1.4%). Of the 213 patients in the registry, 204 (95.8%) are receiving medications that were started on or after the study entry date (Table 2). These concomitant medications included corticosteroids (67.1%), vitamin D and vitamin D analogs (53.5%), ACE inhibitors (26.8%), calcium (21.6%), proton pump inhibitors (10.8%) and osmotic laxatives (6.1%). The concomitant corticosteroids included deflazacort (30.5%), prednisolone (19.7%) and prednisone (19.7%).
| Concomitant medication | Patients in the evaluable population n = 213 |
|---|---|
| Patients receiving concomitant medications, n (%) | 204 (95.8) |
| Glucocorticoids | 143 (67.1) |
| Deflazacort | 65 (30.5) |
| Prednisolone | 42 (19.7) |
| Prednisone | 42 (19.7) |
| Vitamin D and vitamin D analogs | 114 (53.5) |
| Calcifediol | 8 (3.8) |
| Calcitriol | 1 (0.5) |
| Cholecalciferol | 53 (24.9) |
| Ergocalciferol | 5 (2.3) |
| Vitamin D (not otherwise specified) | 48 (22.5) |
| ACE inhibitors | 57 (26.8) |
| Enalapril maleate | 13 (6.1) |
| Perindopril | 15 (7.0) |
| Perindopril arginine | 22 (10.3) |
| Perindopril erbumine | 2 (0.9) |
| Ramipril | 6 (2.8) |
| Calcium | 46 (21.6) |
| Calcium | 24 (11.3) |
| Calcium carbonate | 20 (9.4) |
| Calcium citrate | 1 (0.5) |
| Calcium phosphate | 1 (0.5) |
| Proton pump inhibitors | 23 (10.8) |
| Esomeprazole magnesium | 2 (0.9) |
| Lansoprazole | 6 (2.8) |
| Omeprazole | 12 (5.6) |
| Omeprazole magnesium | 1 (0.5) |
| Pantoprazole sodium sesquihydrate | 1 (0.5) |
| Rabeprazole sodium | 1 (0.5) |
| Osmotic laxatives | 13 (6.1) |
| Lactitol | 1 (0.5) |
| Macrogol | 1 (0.5) |
| Macrogol 3350; potassium chloride; sodium bicarbonate | 2 (0.9) |
| Macrogol 4000 | 5 (2.3) |
| Macrogol; potassium chloride; sodium bicarbonate; sodium chloride | 1 (0.5) |
| Mannitol | 1 (0.5) |
| Potassium; sodium bicarbonate | 2 (0.9) |
| Calcium, combinations with vitamin D and/or other drugs | 11 (5.2) |
Concomitant medications were coded using the WHO Drug Dictionary (September 2017). Concomitant medications are defined as any medications that patients started on or after the study treatment start date. Patients may have more than one medication per ATC level three category and preferred term. At each level of patient summarization, a patient is counted once if the patient reported having one or more medications.
ACE: Angiotensin-converting enzyme; ATC: Anatomical therapeutic chemical.
Ataluren use
The 213 patients in the registry used ataluren for a mean (SD) duration of 639.0 (362.9) days or 372.6 patient-years while enrolled in the registry (Table 3).
| Treatment duration | Patients in the evaluable population n = 213 |
|---|---|
| Ataluren use duration (days†) | |
| n | 213 |
| Mean (SD) | 639.0 (362.9) |
| 95% CI | 590.0, 688.0 |
| Median | 617.0 |
| Min, max | 5.0, 1453 |
| Ataluren use duration (patient-years‡) | 372.6 |
| Categories of ataluren use duration in days, n (%) | |
| 1–168 | 14 (6.6) |
| 169–336 | 44 (20.7) |
| 337–504 | 38 (17.8) |
| 505–672 | 17 (8.0) |
| 673–840 | 25 (11.7) |
| 841–1008 | 37 (17.4) |
| 1009–1176 | 23 (10.8) |
| 1177–1344 | 12 (5.6) |
| 1345–1512 | 3 (1.4) |
| >1512 | 0 (0.0) |
†
Calculated using the formula: ataluren use duration = (ataluren end date – ataluren start date + 1). The treatment start date is defined as the date of the patient’s first visit in the study, including ataluren use in retrospective visits. The treatment end date is defined as the last available ataluren stop date or the data cut-off date, whichever occurs first.
‡
Calculated by dividing the total ataluren use in days from all evaluable patients by 365.25.
SD: Standard deviation.
Discussion
The STRIDE Registry constitutes the first drug registry for patients with DMD, and was designed to provide real-world evidence on long-term outcomes for patients with nmDMD receiving ataluren during routine clinical practice. Here, we have described patient demographics, characteristics and symptoms, as well as ataluren duration and use of prior and concomitant medications in the STRIDE Registry, as of the cut-off date of 9 July 2018.
Previously, the CARE-NMD project evaluated the care practices for 1062 patients with DMD in seven European countries using a cross-sectional survey of patients [18]. Patients had a mean (SD) age of 13.0 (7.2) years at questionnaire completion. Results from this survey showed that the mean (SD) patient age at DMD diagnosis by muscle biopsy or genetic testing was 4.3 (2.5) years (range of the means across countries, 3.7–6.4 years), and the mean (SD) time from first symptoms to this diagnosis was 1.3 (1.8) years (range of the means across countries, 0.8–2.0 years). In the STRIDE Registry, the mean (SD) ages of patients at muscle biopsy and at genetic diagnosis were 4.5 (2.5) years and 5.2 (2.9) years, respectively; the time from first symptoms to muscle biopsy and from first symptoms to genetic diagnosis was 1.6 (1.9) years and 2.4 (2.4) years, respectively. Comparisons of diagnosis times between patients surveyed by the CARE-NMD project and patients in the STRIDE Registry cannot be made accurately because the CARE-NMD survey included all patients with DMD, the majority of whom can be readily diagnosed, rather than exclusively patients with nmDMD as in the STRIDE Registry, and only 71.6% of patients evaluated in CARE-NMD reported having genetic testing for DMD mutations. Moreover, the proportion of patients from different countries differed between the two studies; the STRIDE Registry included large proportions of patients from France (25.4%) and Italy (23.0%), whereas, the CARE-NMD project did not include patients from these countries and included a larger proportion of patients from Germany (39.5%) than did the STRIDE Registry (15.5%). These differences in demographics could affect the diagnosis results reported owing to potential differences in clinical practice between countries. Nevertheless, the age at muscle biopsy of patients in the STRIDE Registry is similar to the age at diagnosis by muscle biopsy or genetic testing of patients surveyed in the CARE-NMD project. Results from a separate study of 384 patients with DMD in Italy showed that the mean age at diagnosis for these patients was 3.4 years, and the mean time between first symptoms and diagnosis was 1.0 years [19]. Of the 382 patients in whom diagnosis of DMD was genetically confirmed, 109 (28.5%) were genetically diagnosed before muscle biopsy [19]. Therefore, again, these figures cannot be easily compared with those from the STRIDE Registry, but suggest that patients in the STRIDE Registry are diagnosed later than those in the total DMD population. Patients with nmDMD in the STRIDE Registry are likely diagnosed later owing to the sequential genetic testing process for DMD introducing delays in DMD diagnosis. Approximately 80% of patients with DMD have a deletion or duplication of one or more exons in their DMD gene, and can be identified by multiplex ligation-dependent probe amplification or comparative genome hybridization, the widely available recommended testing approaches to use first [20]. The remaining approximately 20% of patients without large deletions or duplications require further genetic testing by Sanger or next-generation sequencing to identify any small mutations including nonsense mutations. After this, if no mutation is identified, a muscle biopsy is taken to determine whether dystrophin is absent to exclude other forms of muscular dystrophy. mRNA analysis should then also be performed to identify any potential intronic mutations, which typically affect <1% of patients with DMD [20]. The resulting longer diagnostic process for patients with nmDMD highlights a potential unmet need for this patient group that should be addressed. However, compared with 5 years ago, next-generation sequencing is now more accessible and less expensive, meaning that performing the second step in the genetic testing process is more feasible now than in recent years [21]. It is noteworthy that although guidelines recommend taking a muscle biopsy only if a genetic diagnosis is inconclusive [20], data from the STRIDE Registry show that a muscle biopsy is routinely performed before genetic confirmation of DMD.
Importantly, nine of the patients with DMD recruited into the registry carried frameshift mutations, and are therefore not expected to benefit from ataluren treatment. The initiation of their ataluren treatment by their treating physician was in error owing to misinterpretation of the genetic laboratory report, which indicated the presence of a premature stop codon, albeit one arising from a frameshift mutation and not a nonsense mutation. These patients with frameshift mutations have since stopped receiving ataluren upon guidance from their treating physician, and will be excluded from the effectiveness analyses. This finding highlights the need for continuous professional development of the medical care team, especially when there are new personalized medications that require interpretation of complex genetic information.
As a rare disease, DMD affects relatively few people; there is therefore little documented information on rare disease epidemiology, particularly for the different genotypic subgroups of patients with DMD, and it is often difficult to recruit large numbers of patients into clinical trials [22]. Furthermore, patients with rare diseases tend to have more heterogeneous clinical features than patients with more common diseases [22]. Registries such as the STRIDE Registry are therefore important for gathering and integrating data on patients with rare diseases, and can be used for determining real-world outcomes for diverse purposes, including the natural history of a disease and the effectiveness and safety of therapies.
As a real-world study, STRIDE contains a more heterogeneous population of patients than that in a randomized clinical trial, which could affect the data collected. For example, the use of corticosteroids in this study (67% at study entry and 89.2% during the study and before data cut-off) is less than the 100% use required in the Phase III ACT DMD trial of ataluren. As a consequence, there will be a wider range of ages and ambulatory ability in the STRIDE Registry than in clinical trials, making it more difficult to draw conclusions. However, this also means that data from the STRIDE Registry will be more representative of real-world patients’ experiences. This will therefore help to progress our understanding of the treatment effects of ataluren and provide real-world data on the care that patients receive, as well as more information on nonambulatory patients, who are often omitted from clinical trials. The collaborative Trajectory Analysis Project, which accesses and analyzes data from patients in natural history studies [23], and the CINRG DNHS, which was a prospective longitudinal study of patients with DMD followed between 2006 and 2016 at 23 world-wide centers part of the CINRG academic clinical trial network [24,25], could be useful for placing real-word data from the STRIDE Registry in context. It is noteworthy that the mean (SD) ataluren use duration for patients while enrolled in the STRIDE Registry was 639.0 (362.9) days, which is longer than the 336 days (48 weeks) of ataluren use in each of the Phase IIb and ACT DMD Phase III clinical trials of ataluren [11,12]. In addition, because some patients were enrolled in the STRIDE Registry after being involved in a clinical trial of ataluren, the actual mean ataluren use duration for patients in the registry would be longer than 639 days. The STRIDE Registry therefore provides the opportunity to follow-up patients over a longer period than can be afforded by clinical studies thus far, which are designed to show the benefit of treatment in delaying disease progression and loss of muscle function. Furthermore, data from the STRIDE Registry will provide additional characterization and quantification of AEs that may have a low incidence or a long latency period after exposure to ataluren, or that may be more likely to occur outside the controlled setting of a clinical trial.
Conclusion
In summary, STRIDE constitutes the first drug registry for patients with DMD and represents the largest real-world study of patients with nmDMD to date. The STRIDE Registry has been established to promote collection of real-world evidence on ataluren use in patients with nmDMD and improve understanding of the long-term safety and effectiveness of ataluren. A manuscript describing the safety and real-world effectiveness of ataluren in the STRIDE Registry, as of the cut-off date of 9 July 2018, will be published separately in due course. In this publication, the effectiveness of ataluren will be assessed by comparing results from propensity-score-matched patient populations from the STRIDE Registry and the CINRG DNHS.
•
Duchenne muscular dystrophy (DMD) is a rare, progressive, genetic, neuromuscular disorder.
•
Approximately 10–15% of patients have a nonsense mutation (nm) in the DMD gene coding for the dystrophin protein.
•
Ataluren promotes readthrough of an in-frame premature stop codon, enabling the production of full-length dystrophin.
•
Strategic Targeting of Registries and International Database of Excellence (STRIDE) is the largest real-world study of patients with nmDMD to date.
•
STRIDE is an ongoing registry of patients receiving ataluren in clinical practice.
•
As of 9 July 2018, 213 boys have been enrolled in STRIDE from 11 countries.
•
Mean age was 2.7 and 5.2 years at first symptoms and at genetic diagnosis, respectively.
•
Mean age at first captured visit in STRIDE (treatment start date) was 9.8 years.
•
Corticosteroids were used by 190 patients (89.2%) during the study and before data cut-off.
•
Total mean exposure to ataluren within the registry was 639.0 days.
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: Supplementary Material
Author contributions
F Muntoni, I Desguerre, M Guglieri, A Nascimento Osorio, J Kirschner, M Tulinius and E Mercuri are members of the STRIDE Registry Steering Committee and have enrolled patients at their study site. G Elfring, C Werner, T Schilling, P Trifillis, O Zhang, A Delage and CL Santos were involved in the design of the study. F Buccella is a member of the STRIDE Registry Steering Committee and, as an expert patient advocate, he reviewed the manuscript and approved the final version for submission. F Muntoni, I Desguerre, M Guglieri, A Nascimento Osorio, J Kirschner, M Tulinius, F Buccella, G Elfring, C Werner, T Schilling, P Trifillis, O Zhang, A Delage, CL Santos and E Mercuri reviewed data, drafted and critically reviewed the manuscript and approved the final version for submission.
Acknowledgments
We thank the patients and their families for their participation in this study. We also thank the individuals involved in the conduct of this study and the collection of data, particularly the STRIDE principal investigators and study coordinators.
Financial & competing interests disclosure
This work was supported by PTC Therapeutics. The STRIDE Registry is sponsored by PTC Therapeutics. F Muntoni has received consultancy fees from Akashi Therapeutics, Biogen, BioMarin, Catabasis, Italfarmaco, Pfizer, PTC Therapeutics, Roche, Sarepta Therapeutics and Tivorsan Pharmaceuticals, and is supported by the National Institute of Health Research Biomedical Research Centre at Great Ormond Street Hospital for Children National Health Service Foundation Trust and University College London. I Desguerre has received consultancy fees from AveXis, Biogen, BioMarin and PTC Therapeutics. M Guglieri is a site Principal Investigator for the PTC Therapeutics extension study of ataluren in DMD, has acted as a consultant/advisory board member for BioMarin, Capricor, Debiopharm, Marathon Pharmaceuticals and PTC Therapeutics, and is the chief medical coordinator of the FOR DMD study and study with vamorolone in DMD. AN Osorio has received speaker and consultancy fees from PTC Therapeutics, and is an investigator on clinical trials sponsored by Biogen, F. Hoffmann-La Roche, Italfarmaco, Sarepta Therapeutics, and TAMDMD. J Kirschner has acted as a consultant for AveXis, Biogen, Ionis Pharmaceuticals, PTC Therapeutics and Roche, and has received research support for taking part in clinical research from Biogen, BioMarin, GlaxoSmithKline, Ionis Pharmaceuticals, Novartis, PTC Therapeutics, Roche, Santhera Pharmaceuticals and Trophos. M Tulinius has received lecture fees from Biogen and PTC Therapeutics, and has acted as a consultant on DMD clinical trials for BioMarin, PTC Therapeutics, ReveraGen BioPharma, and Sarepta Therapeutics, and as an advisory board member for AveXis, Biogen and PTC Therapeutics. F Buccella has received consultancy fees from PTC Therapeutics, Santhera Pharmaceuticals, and Sarepta Therapeutics. T Schilling, P Trifillis and CL Santos are full-time employees of PTC Therapeutics, Inc. C Werner is a full-time employee of PTC Therapeutics Germany GmbH, and A Delage is a full-time employee of PTC Therapeutics Switzerland GmbH. G Elfring is a consultant for PTC Therapeutics. E Mercuri has acted as an advisory board member for AveXis, Biogen, BioMarin, Bristol-Myers Squibb, Ionis Pharmaceuticals, Italfarmaco, Prosensa, PTC Therapeutics, Roche, Santhera Pharmaceuticals, Sarepta Therapeutics and Summit Therapeutics. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Medical writing and editorial support were provided by T Ellison, PhD, of PharmaGenesis London, London, UK, and were funded by PTC Therapeutics.
Ethical conduct of research
Before any patient is enrolled and study-related data are collected, ethics committee approval of the protocol, informed consent form and all patient enrollment materials are obtained in each country and for each site, as applicable. The study is conducted in accordance with the ethical principles outlined in the Declaration of Helsinki, applicable privacy laws and local regulations for each participating site, as well as with the Guidelines for Good Pharmacoepidemiology Practices.
Data sharing statement
Individual, patient data will not be made publicly available. The study protocol is available indefinitely to interested parties on request.
Open access
This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
Supplementary Material
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
1.
Ellis JA, Vroom E, Muntoni F. 195th ENMC International Workshop: newborn screening for Duchenne muscular dystrophy December 14–16, 2012, Naarden, The Netherlands. Neuromuscul. Disord. 23(8), 682–689 (2013).
2.
Bushby K, Finkel R, Birnkrant DJ et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 9(1), 77–93 (2010).
3.
Birnkrant DJ, Bushby K, Bann CM et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 17, 251–267 (2018).
• One article in a three-part overview of the 2018 standard of care guidelines for the management of patients with Duchenne muscular dystrophy (DMD).
4.
Pichavant C, Aartsma-Rus A, Clemens PR et al. Current status of pharmaceutical and genetic therapeutic approaches to treat DMD. Mol. Ther. 19(5), 830–840 (2011).
5.
Bladen CL, Salgado D, Monges S et al. The TREAT-NMD DMD Global Database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum. Mutat. 36(4), 395–402 (2015).
6.
Laing NG, Davis MR, Bayley K, Fletcher S, Wilton SD. Molecular diagnosis of duchenne muscular dystrophy: past, present and future in relation to implementing therapies. Clin. Biochem. Rev. 32(3), 129–134 (2011).
7.
Bello L, Pegoraro E. Genetic diagnosis as a tool for personalized treatment of Duchenne muscular dystrophy. Acta. Myol. 35(3), 122–127 (2016).
8.
European Medicines Agency. Translarna™ (ataluren) summary of product characteristics (2018). www.ema.europa.eu/documents/product-information/translarna-epar-product-information_en.pdf
9.
Peltz SW, Morsy M, Welch EM, Jacobson A. Ataluren as an agent for therapeutic nonsense suppression. Annu. Rev. Med. 64, 407–425 (2013).
10.
Roy B, Friesen WJ, Tomizawa Y et al. Ataluren stimulates ribosomal selection of near-cognate tRNAs to promote nonsense suppression. Proc. Natl Acad. Sci. USA 113(44), 12508–12513 (2016).
11.
Bushby K, Finkel R, Wong B et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve 50(4), 477–487 (2014).
•• Describes the safety and efficacy results of ataluren in a Phase IIb randomized, double-blind, placebo-controlled trial.
12.
Mcdonald CM, Campbell C, Torricelli RE et al. Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): a multicentre, randomised, double-blind, placebo-controlled, Phase III trial. Lancet 390(10101), 1489–1498 (2017).
•• Describes the safety and efficacy results of ataluren in a Phase III randomized, double-blind, placebo-controlled trial.
13.
European Medicines Agency. Summary of the risk management plan (RMP) for Translarna (ataluren) (2014). www.ema.europa.eu/en/documents/rmp-summary/translarna-epar-risk-management-plan-summary_en.pdf
14.
Birnkrant DJ, Bushby K, Bann CM et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. 17, 347–361 (2018).
• One article in a three-part overview of the 2018 standard of care guidelines for the management of patients with DMD.
15.
Birnkrant DJ, Bushby K, Bann CM et al. Diagnosis and management of Duchenne muscular dystrophy, part 3: primary care, emergency management, psychosocial care, and transitions of care across the lifespan. Lancet Neurol. 17, 445–455 (2018).
• One article in a three-part overview of the 2018 standard of care guidelines for the management of patients with DMD.
16.
Mazzone ES, Messina S, Vasco G et al. Reliability of the North Star Ambulatory Assessment in a multicentric setting. Neuromuscul. Disord. 19(7), 458–461 (2009).
17.
Mayhew A, Mazzone ES, Eagle M et al. Development of the Performance of the Upper Limb module for Duchenne muscular dystrophy. Dev. Med. Child Neurol. 55(11), 1038–1045 (2013).
18.
Vry J, Gramsch K, Rodger S et al. European cross-sectional survey of current care practices for Duchenne muscular dystrophy reveals regional and age-dependent differences. J. Neuromuscul. Dis. 3(4), 517–527 (2016).
•• Describes patient demographic results from the CARE-NMD project, which evaluated the care practices for 1062 patients with DMD in seven European countries using a cross-sectional survey of patients.
19.
D'Amico A, Catteruccia M, Baranello G et al. Diagnosis of Duchenne muscular dystrophy in Italy in the last decade: critical issues and areas for improvements. Neuromuscul. Disord. 27(5), 447–451 (2017).
•• Describes patient demographic results from a retrospective study investigating the age at diagnosis of DMD in Italy.
20.
Aartsma-Rus A, Ginjaar IB, Bushby K. The importance of genetic diagnosis for Duchenne muscular dystrophy. J. Med. Genet. 53(3), 145–151 (2016).
21.
Fernandez-Marmiesse A, Gouveia S, Couce ML. NGS technologies as a turning point in rare disease research, diagnosis and treatment. Curr. Med. Chem. 25(3), 404–432 (2018).
22.
Martins AM, Kyosen SO. The importance of patient registries for rare diseases. Expert Opin. Orphan Drugs 1(10), 769–772 (2013).
• Describes the heterogeneity of rare diseases and the importance of gathering information in registries about patients with rare diseases.
23.
Collaborative Trajectory Analysis Project. Enabling the right trial design, the first time (2016). http://ctap-duchenne.org/assets/files/cTAP-Overview_2016.pdf
24.
Mcdonald CM, Henricson EK, Abresch RT et al. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: a prospective cohort study. Lancet 391(10119), 451–461 (2018).
25.
Mcdonald CM, Henricson EK, Abresch RT et al. The cooperative international neuromuscular research group Duchenne natural history study–a longitudinal investigation in the era of glucocorticoid therapy: design of protocol and the methods used. Muscle Nerve 48(1), 32–54 (2013).
Information & Authors
Information
Published In
Pages: 1187 - 1200
PubMed: 31414621
Copyright
© 2019 Muntoni et al. This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 Unported License
History
Received: 3 July 2019
Accepted: 29 July 2019
Published online: 15 August 2019
Keywords:
Topics
Authors
Metrics & Citations
Metrics
Article Usage
Article usage data only available from February 2023. Historical article usage data, showing the number of article downloads, is available upon request.
Citations
How to Cite
Ataluren use in patients with nonsense mutation Duchenne muscular dystrophy: patient demographics and characteristics from the STRIDE Registry. (2019) Journal of Comparative Effectiveness Research. DOI: 10.2217/cer-2019-0086
Export citation
Select the citation format you wish to export for this article or chapter.
Citing Literature
- Richard B. Silverman, Fengtian Xue, José I. Juncosa, Drugs Interacting with Nucleic Acids, The Organic Chemistry of Drug Design and Drug Action, 10.1016/B978-0-443-15862-9.00009-6, (343-396), (2027).
- Elena Cannone, Martina La Spina, Barbara Gnutti, Chiara Tesoriero, Silvia Castagnaro, Chiara Tobia, Luca La Via, Patrizia Sabatelli, Cesare Faldini, Giuseppe Fiume, Andrea Vettori, Dario Finazzi, Massimo Gennarelli, Chiara Magri, Marco Schiavone, Integrated RNA sequencing and in vivo biosensor imaging define the early pathogenic cascade of Duchenne muscular dystrophy, Translational Research, 10.1016/j.trsl.2026.06.007, 295, (31-49), (2026).
- N. V. Vyalova, I. P. Piotrovich, N. V. Sikora, A. S. Skretnev, V. O. Kovalenko, M. V. Eliseeva, DUCHENNE MYODYSTROPHY, Transbaikalian Medical Bulletin, 10.52485/19986173_2025_4_157, 4, (157-164), (2026).
- Zakaria Rostamitehrani, Rida Javed, Linda Popplewell, Dystrophin Restorative and Compensatory Gene Addition Therapies for Duchenne Muscular Dystrophy: Could CRISPRa Provide a Realistic Alternative?, Muscles, 10.3390/muscles4040052, 4, 4, (52), (2025).
- Dmitry Vlodavets, Shiwen Wu, Anna Kostera-Pruszczyk, Jong-Hee Chae, Sheffali Gulati, Jin-Hong Shin, Michelle Lorentzos, Anita Cairns, Yuh-Jyh Jong, Hirofumi Komaki, Jeffrey Statland, Alexandra Prufer de Queiroz Campos Araujo, Juliana Gurgel-Giannetti, Yasuhiro Takeshima, Rosa E Escobar Cedillo, Peter Karachunski, Kazuhiro Haginoya, Vinay Penematsa, Connie Chou, Paula Williams, Christian Werner, Craig M McDonald, Confirmatory long-term efficacy and safety results of ataluren in patients with nmDMD from Study 041, an international, randomized, double-blind, placebo-controlled, Phase III trial, Journal of Comparative Effectiveness Research, 10.57264/cer-2024-0238, 14, 10, (2025).
- Mohammad Sawahreh, Fatima Al-Maadid, Khalid Omer Ibrahim, Tawfeg Ben Omran, Mahmoud Fawzi Osman, Patient demographics, clinical characteristics and genetic mutations of DMD and BMD patients in Qatar Epidemiological and genetic profile of Duchenne muscular dystrophy and Becker muscular dystrophy patients in Qatar: a retrospective cohort study, Frontiers in Pediatrics, 10.3389/fped.2025.1569505, 13, (2025).
- Mushtaha Ahmad, Alaa ElRasoul, Raneem Sedayou, Mohammed Tamboosi, Hanan Mahroos, Shaimaa Alrashed, Mariam Tunkar, Faisal Alzahrani, Mohammed Alharbi, Mona Aljehani, Mousa Alahmari, Khalid Alqarni, Maha Gashlan, Berna Seker Yilmaz, Nahla M. Alshaikh, Safety and effectiveness of ataluren in patients with Duchenne muscular dystrophy: single-center experience from Saudi Arabia, Journal of International Medical Research, 10.1177/03000605241305252, 52, 12, (2024).
- Tanja Golli, Lenka Juříková, Thomas Sejersen, Craig Dixon, The role of ataluren in the treatment of ambulatory and non-ambulatory children with nonsense mutation duchenne muscular dystrophy - a consensus derived using a modified Delphi methodology in Eastern Europe, Greece, Israel and Sweden, BMC Neurology, 10.1186/s12883-024-03570-x, 24, 1, (2024).
- Shan Li, Juan Li, Wenjing Shi, Ziyan Nie, Shasha Zhang, Fengdie Ma, Jun Hu, Jianjun Chen, Peiqiang Li, Xiaodong Xie, Pharmaceuticals Promoting Premature Termination Codon Readthrough: Progress in Development, Biomolecules, 10.3390/biom13060988, 13, 6, (988), (2023).
- Eugenio Mercuri, Andrés Nascimento Osorio, Francesco Muntoni, Filippo Buccella, Isabelle Desguerre, Janbernd Kirschner, Már Tulinius, Maria Bernadete Dutra de Resende, Lauren P. Morgenroth, Heather Gordish-Dressman, Shelley Johnson, Allan Kristensen, Christian Werner, Panayiota Trifillis, Erik K. Henricson, Craig M. McDonald, Safety and effectiveness of ataluren in patients with nonsense mutation DMD in the STRIDE Registry compared with the CINRG Duchenne Natural History Study (2015–2022): 2022 interim analysis, Journal of Neurology, 10.1007/s00415-023-11687-1, 270, 8, (3896-3913), (2023).