Network connectivity, between-study heterogeneity and timepoint challenges in generalized myasthenia gravis: a feasibility assessment of indirect treatment comparisons
Publication: Journal of Comparative Effectiveness Research
Abstract
Aim: We performed a feasibility assessment to systematically evaluate randomized controlled trials (RCTs) for generalized myasthenia gravis (gMG) treatments. The goal was to identify the advantages and disadvantages of different indirect treatment comparison (ITC) methods. Materials & methods: A systematic literature review was conducted to identify relevant gMG RCTs for ITCs. The feasibility of ITCs was assessed by comparing design (including study duration and dosing schedules), population and outcome characteristics of retrieved trials, investigating network connectivity and considering appropriate ITC methods to address identified challenges. Results: The feasibility assessment considered 15 relevant RCTs for gMG treatments. Several barriers to conducting robust ITCs were identified, including within-trial imbalances in patient characteristics, small trial sizes and cross-trial differences in potential treatment effect modifiers (TEMs; e.g., antibody status, disease duration and prior treatment exposure). Further, heterogeneity in placebo administration characteristics and background therapies, and cross-trial variation in placebo response for key outcomes were noted. Additionally, treatment strategies (i.e., cyclical vs continuous), dosing schedules and outcome assessment timepoints were inconsistent across trials, necessitating careful consideration of methods and timepoints when interpreting outcomes. The findings suggest that ITCs anchored on placebo as a common comparator may be prone to bias, and more than one ITC approach may be necessary. Conclusion: ITC analyses in gMG have inherent challenges related to imbalanced treatment effect modifiers, network connectivity, varying dosing strategies and assessment timepoints. Multiple approaches to ITCs, with careful evaluation of underlying assumptions and limitations, are advised to limit bias and ensure robust comparative efficacy estimates are available to decision makers.
Plain language summary: Investigating differences between clinical trials of treatments for generalized myasthenia gravis to identify appropriate indirect treatment comparison methods
What is this article about?
Generalized myasthenia gravis (gMG) is a rare condition where communication is disrupted between nerves and muscles impacting the ability to see clearly, eat, breathe and do daily activities. There are multiple treatments available for gMG that target this disruption, but their effectiveness has not been directly compared in a clinical trial. In this situation, analyses known as indirect treatment comparisons (ITCs) can inform how gMG treatments compare with each other. There are multiple ITC methods, and they have different requirements, so it is important to closely examine the clinical trials for gMG treatments to see if there are differences between them that could lead to unreliable results. In this work, we examined 15 clinical trials for gMG treatments to identify which ITC methods were most appropriate to generate reliable comparisons.
What were the results?
We identified several barriers to conducting robust ITCs, including differences in the characteristics of patients included in trials, as well as differences in other treatments patients could be receiving during a trial, the way in which treatments were administered, and when patients were evaluated during the trials to determine how effective the treatments were.
What do the results mean?
It is important to use multiple ITC methods to understand how gMG treatment compare with each other, and to carefully evaluate the underlying assumptions and limitations of these methods.
Generalized myasthenia gravis (gMG) is a rare autoimmune disorder involving disruption of neuromuscular junction (NMJ) signaling, which manifests as skeletal muscle weakness and fatigue. Autoantibodies to acetylcholine receptors (AChR) at the NMJ are commonly present in gMG, with other targets including muscle-specific kinase (MuSK), and low-density lipoprotein receptor-related protein 4 (LRP4). As a chronic systemic condition, gMG has a substantial clinical, social and economic burden [1–4].
Standard of care (SOC) for gMG includes acetylcholinesterase inhibitors, corticosteroids and nonsteroidal immunosuppressant therapies (NSISTs) such as azathioprine, mycophenolate and tacrolimus either as monotherapy or in combination [5,6]. Newer targeted immunotherapies added to SOC include neonatal Fc receptor (FcRn) and C5 complement inhibitors, which act on immune pathways to constrain disease severity. Other add-on immunotherapies include rituximab (RTX; targeted to CD20) and non-acute intravenous immunoglobulin (IVIg) [7–9].
Registration randomized controlled trials (RCTs) for newer targeted immunotherapies (efgartigimod: ADAPT [10]; rozanolixizumab: MycarinG [11]; nipocalimab: Vivacity-MG3 [12]; eculizumab: REGAIN [13]; ravulizumab: CHAMPION-MG [14]; and zilucoplan: RAISE [15]) administered in combination with SOC have demonstrated positive results versus placebo plus SOC. Although RCT evidence is the gold standard for assessing comparative efficacy, there are currently no head-to-head RCTs for add-on immunotherapies in gMG. Therefore, indirect treatment comparisons (ITCs) are potentially a valuable tool for understanding the relative effectiveness of gMG therapies [16,17].
ITCs leveraging RCT data can be grouped into three categories. Network meta-analysis (NMA) allows simultaneous comparison of multiple treatments in a connected evidence network based on shared treatment arms, only requires aggregate data [16,18]. A Bucher ITC may be considered as a simplified NMA comparing only two treatments through a common comparator [19]. A second category of ITCs uses a combination of aggregate data and individual patient data (IPD) from RCTs, allowing adjustment for population differences between trials. Matching-adjusted indirect comparisons (MAICs) and simulated treatment comparisons (STCs) compare two treatments where IPD are available for a trial of one treatment [17,20]. Anchored MAICs/STCs compare treatments through a common comparator, whereas unanchored MAICs/STCs do not. Finally, propensity score matching/reweighting and regression methods can be used to indirectly compare treatments for which IPD data are available for all relevant RCTs. Approaches to ITC also differ in their underlying assumptions. For example, a key assumption underlying NMAs and Bucher ITCs is exchangeability of trial participants [21].
In gMG, ITCs have been submitted to regulatory and health technology assessment (HTA) agencies and are increasingly being published. However, agencies have expressed dissatisfaction with submitted ITCs for several reasons, including the lack of justification for selected methods and inconsistency in results [22–26]. Specific limitations included the use of ITC approaches that did not account for variation in dosing regimens, trial eligibility criteria and baseline characteristics; bias introduced by choice of timepoints; inclusion of trials with small sample sizes; and poorly explained covariate selection in MAICs/STCs. Similar issues have been raised for published ITCs [27].
The selection of appropriate ITC methods should be informed by the available evidence and tailored to the unique challenges encountered in a particular disease and treatment space. A comprehensive assessment of the characteristics (e.g., study design, population and outcomes) of the trials informing the evidence base for a condition and treatments of interest is required to identify sources of heterogeneity that could bias an ITC and thereby undermine the validity of results [21,28–31]. In particular, it is important to consider any heterogeneity in placebo arm characteristics (e.g., background therapies) and cross-trial variation in placebo response for key outcomes (e.g., due to expectation bias for some trials). Differences in placebo response across trials may reflect imbalances in reported (e.g., age, sex, baseline disease severity and antibody status) and unreported treatment effect modifiers (TEMs), and thereby introduce bias in anchored ITCs such as NMA if not properly accounted for [32,33]. Placebo response may also be influenced by route of administration [34–36] as well as by class, frequency and dosing of background SOC therapy. Assessing the feasibility of ITC is important to understand any key assumptions and limitations of the considered methods so they can be communicated appropriately and addressed where possible [37].
Given the previously identified concerns with ITCs in the gMG treatment space, we conducted an in-depth systematic feasibility assessment in line with available guidance from HTA agencies and health economics bodies [21,28–31] to evaluate currently available trials for gMG treatments, with the goal of identifying which ITC methods are most appropriate to generate reliable comparative efficacy estimates.
Materials & methods
Overview
A systematic literature review (SLR) to identify clinical trials reporting on the efficacy and safety of systemic treatments for patients with gMG was conducted. Detailed SLR methods are provided in the Supplementary Materials. The SLR was designed and reported in accordance with the Preferred Reporting Items for Systematic Literature Reviews and Meta-Analysis (PRISMA) guidelines [38]. An SLR protocol and an ITC protocol were developed a priori. IPD-only analyses were assumed to be infeasible because IPD from RCTs for more than one treatment are seldom available to the same investigators. Hence, ITC feasibility assessment was restricted to NMAs and MAICs/STCs.
SLR: search strategy
We searched biomedical databases, recent neurology conference proceedings (i.e., American Academy of Neurology, American Association of Neuromuscular and Electrodiagnostic Medicine, and European Academy of Neurology from 2021 to 2023), regulatory and HTA agency websites, and ClinicalTrials.gov. Additionally, bibliographies of relevant SLR and ITC publications were checked for relevant trials.
SLR: study selection & data extraction
Two independent reviewers screened citations according to prespecified eligibility (PICOS) criteria. The eligible population was patients diagnosed with gMG. Interventions of interest were SOC therapies, add-on immunotherapies and thymectomy. Outcomes of interest included the efficacy outcomes Myasthenia Gravis Activities of Daily Living (MG-ADL) and Quantitative Myasthenia Gravis (QMG). Only clinical trials published in English from 2003 onward (i.e., in the last 20 years) were included as older trials may have limited relevance to current management of gMG.
In alignment with EU guidance, only RCTs were considered for the ITC feasibility assessment as they are the preferred data source for ITCs [21]. The evidence base for the feasibility assessment was comprised of publications of phase II or III RCTs studying US FDA and EMA approved dosing regimens (and routes of administration) of newer targeted immunotherapies added to SOC for gMG, off-label add-on immunotherapies to SOC recommended in published national or international gMG treatment guidelines (i.e., rituximab and non-acute IVIg) and nipocalimab (only the Vivacity-MG3 phase III trial was included) [8,9]. Although results for the nipocalimab Vivacity-MG3 phase III RCT were not published at the time of the SLR searches, this trial was included because a publication became available during preparation of this manuscript [12]. A phase II RCT by Wolfe et al. (2002) was also considered for the feasibility assessment [39]. Although this trial was outside the SLR date limit, it was identified as the only RCT reporting MG-ADL change from baseline (CFB) data for non-acute IVIg and had been included in a previous ITC [22].
Data extraction was performed by a single reviewer and independently assessed for accuracy and completeness by a second reviewer. Extracted data included study, patient and outcome characteristics.
Feasibility assessment
The following trial characteristics were assessed to inform the feasibility of ITCs: study design characteristics, treatment characteristics, trial eligibility criteria, patient characteristics at baseline and outcome characteristics. Heterogeneity in baseline risk (i.e., placebo arm outcomes) was also assessed. Outcomes of interest were MG-ADL and QMG CFB and response (i.e., proportion reaching a within-person meaningful change threshold) as these were the most commonly reported primary or key secondary efficacy outcomes in the RCTs.
We conducted the ITC feasibility assessment in three steps: investigation of reported trial information to identify substantial within- and cross-trial differences; assessment of network connectivity to understand whether anchored ITC methods such as NMA were appropriate; and synthesis of findings and identification of appropriate ITC methods. Our approach was based on guidance from Cochrane, the EU, NICE and the International Society for Pharmacoeconomics and Outcomes Research (ISPOR) [21,28–31,37,40–42].
We first qualitatively assessed whether clinically important differences in patient characteristics at baseline (e.g., age, sex, baseline MG-ADL score, baseline QMG score, prior corticosteroid exposure, prior NSIST exposure) were present across treatment groups within each trial. Potential imbalances were identified by calculating the standardized mean difference (SMD); an SMD > 0.2 was considered a substantial difference [43] and SMDs between 0.1 and 0.2 were also noted. We also investigated whether any patient characteristics were adjusted for in the models used to calculate MG-ADL and QMG outcomes in each trial, as this would help mitigate potential bias due to within-trial imbalances. Next, we assessed between-trial differences in study design characteristics (including treatment details), population characteristics (based on trial eligibility criteria and summary measures of baseline characteristics), and outcome characteristics (definitions and assessment timepoints). We examined reported subgroup analyses for six registration phase III RCTs to identify potential TEMs. This approach helped contextualize the potential importance of any cross-trial differences in baseline patient characteristics. A summary of reported subgroup analyses and the criterion used to identify potential TEMs is provided in Supplementary Table 1. Treatment schedules, timepoints at which CFB outcomes for MG-ADL and QMG were assessed relative to baseline, and the minimum point improvements (MPIs) (i.e., within-person meaningful change thresholds) used to define MG-ADL and QMG responders, were compared across included RCTs.
Network connectivity was assessed by examining overlap in the treatment arms of included RCTs. Network diagrams were used to visualize the connectivity of treatments. The availability of a connected network and its structure are important determinants toward the selection of ITC methodology [30,31]. A generalized network diagram was constructed based on included RCTs independent of outcome availability and potential issues with the validity of common comparators. The characteristics of the placebo arms of the different trials, such as route of administration, as well as placebo response for MG-ADL and QMG outcomes were compared with identify any cross-trial differences that could introduce bias in an anchored ITC.
Findings from these assessments were synthesized and considered along with trial data availability (i.e., aggregate data vs IPD) to identify: appropriate ITC methods; a base case analysis for each method and potential sensitivity analyses; and limitations of each method.
Results
Systematic literature review
Detailed SLR results are available in the Supplementary Materials. In total, 186 records representing 47 unique trials met the SLR inclusion criteria. Of the 47 trials, 32 were RCTs, seven were open-label extensions of included RCTs and eight were single-arm trials. Fifteen RCTs (including Vivacity-MG3 and Wolfe et al. [2002]) studied add-on immunotherapies and were included in the ITC feasibility assessment. A description of the 19 RCTs captured via the SLR but excluded from the feasibility assessment is provided in the Supplementary Materials. A list of the 15 RCTs included in the feasibility assessment is provided in Supplementary Table 2. A summary of the included trials and their key characteristics is provided in Table 1.
| Trial Name | Phase | Intervention and comparator | Enrolled patients (n) | Treatment administration details | Notable trial eligibility criteria | Ref. |
|---|---|---|---|---|---|---|
| ADAPT | III | Efgartigimod vs placebo | 167 | IV infusion QW for 4-week cycles, repeated as needed depending on clinical response no sooner than 8 weeks after initiation of the previous cycle | • AChR+, MuSK+ or AChR-/MuSK- | [10] |
| ADAPT-SC | III | Efgartigimod IV vs efgartigimod SC | 110 | SC infusion QW for 3 weeks (4 total infusions) | • No criterion for antibody status (all serotypes) | [44] |
| AGRX-113-1602 | II | Efgartigimod vs placebo | 24 | IV infusion QW for 3 weeks | • AChR+ | [45] |
| MycarinG | III | Rozanolixizumab vs placebo | 200 | SC infusion QW for 6 weeks | • AChR+ or MuSK+ | [11] |
| Bril (2021) | II | Rozanolixizumab vs placebo | 43 | SC infusion QW for 3 weeks (period 1) | • AChR+ or MuSK+ | [46] |
| Vivacity-MG3 | III | Nipocalimab vs placebo | 153 | IV infusion Q2W for 24 weeks | • No criterion for antibody status (all serotypes) | [12] |
| REGAIN | III | Eculizumab vs placebo | 126 | IV infusion Q2W for 26 weeks | • AChR+ • Required to have failed on ≥2 IST, or ≥1 IST with IVIg or PLEX given at least four-times per year for 12 months without symptom control | [13] |
| CHAMPION-MG | III | Ravulizumab vs placebo | 175 | IV infusion Q8W for 26 weeks | • AChR+ • Diagnosed with MG ≥6 months before screening | [14] |
| RAISE | III | Zilucoplan vs placebo | 174 | SC infusion QD for 12 weeks | • AChR+ • Excluded patients known to be MuSK+ | [15] |
| Howard (2020) | II | Zilucoplan vs placebo | 45 | SC infusion QD for 12 weeks | • AChR+ • Excluded patients known to be MuSK+ or LRP4+ | [47] |
| RINOMAX | III | Rituximab vs placebo | 47 | IV infusion, single dose | • No criterion for antibody status (all serotypes) • Included patients ≤12 months after onset of gMG | [48] |
| BeatMG | II | Rituximab vs placebo | 52 | IV infusion QW for 4 weeks per cycle, 2 total cycles separated by 6 months | • AChR+ | [49] |
| NCT02473952 | II | Non-acute IVIg vs Placebo | 62 | IV infusion Q3W for 21 weeks | • AChR+ | [50] |
| Bril (2023) | II | Non-acute IVIg vs Placebo | 60 | IV infusion Q3W for 36 weeks | • AChR+ | [51] |
| Wolfe (2002) | II | Non-acute IVIg vs Placebo | 15 | IV infusion at induction and after 3 weeks | • AChR+ | [39] |
AChR+: Anti-acetylcholine receptor antibody-positive; CS: Corticosteroid; gMG: Generalized myasthenia gravis; IS/IM: Immunosuppressant/immunomodulator; IST: Immunosuppressive therapy; ITC: Indirect treatment comparison; IV: Intravenous; IVIg: Intravenous immunoglobulin; LRP4+: Anti-low-density lipoprotein receptor-related protein 4 antibody-positive; MG: Myasthenia gravis; MuSK+: Anti-muscle-specific kinase antibody-positive; PLEX: Plasma exchange; Q2W: Every 2 weeks; Q3W: Every 3 weeks; Q8W: Every 8 weeks; QD: Daily; QW: Weekly; RCT: Randomized controlled trial; SC: Subcutaneous.
Feasibility assessment: assessment of within-trial differences
Within-trial imbalances in baseline patient characteristics were noted for several of the included trials (Figure 1). In general, more extensive imbalances (i.e., SMD > 0.2) were observed for smaller trials (e.g., phase II and non-registration trials such as RINOMAX) compared with larger registration phase III trials. For example, six of the nine baseline characteristics we evaluated for the AGRX-113-1602 phase II efgartigimod RCT were imbalanced based on having an SMD > 0.2, whereas only one of the evaluated characteristics was imbalanced in the ADAPT phase III efgartigimod RCT. Although adjustments for baseline MG-ADL and QMG scores were performed in most included trials, other patient characteristics were generally not adjusted for.
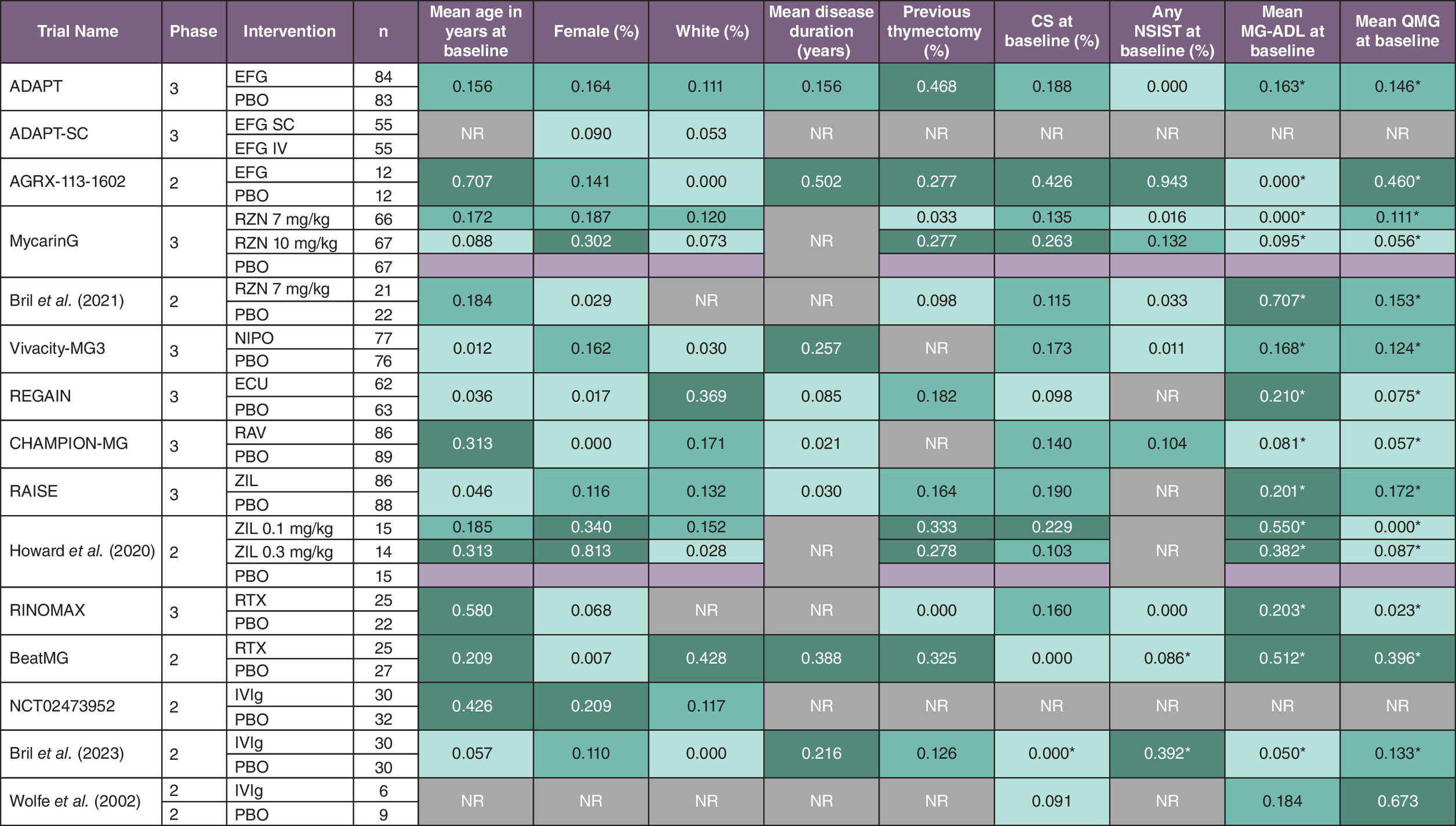
Figure 1. Within-trial imbalances in reported baseline patient characteristics as measured by standardized mean difference and sample sizes among randomized controlled trials included in the indirect treatment comparison feasibility assessment.
Dark green fill indicates an SMD > 0.2 for comparisons with the placebo group, medium green fill indicates an SMD between 0.1 and 0.2 and light green fill indicates an SMD < 0.1.
*Characteristic was adjusted for in models used to calculate MG-ADL and QMG outcomes based on reported trial information.
CS: Corticosteroid; ECU: Eculizumab; EFG: Efgartigimod; ITC: Indirect treatment comparison; IV: Intravenous; IVIg: Intravenous immunoglobulin; MG-ADL: Myasthenia Gravis Activities of daily living; n: Number of patients; NIPO: Nipocalimab; NR: Not reported; NSIST: Nonsteroidal immunosuppressant therapy; PBO: Placebo; QMG: Quantitative Myasthenia Gravis; RAV: Ravulizumab; RCT: Randomized controlled trial; RTX: Rituximab; RZN: Rozanolixizumab; SC: Subcutaneous; SD: Standard deviation; SMD: Standardized mean difference; ZIL: Zilucoplan.
Feasibility assessment: assessment of cross-trial differences
A summary of key trial characteristics is provided in Table 1, patient characteristics at baseline in Table 2 and MG-ADL and QMG assessment details in Table 3. Summaries of general study design characteristics, trial eligibility criteria and outcome characteristics for the 15 included RCTs are available in the Supplementary Materials.
| Trial name | Intervention | Mean age in years at baseline, years (SD) | Female (%) | White (%) | Geographical region | Antibody status | Mean duration of disease (SD) | MGFA | Mean MG-ADL score at baseline (SD) | Mean QMG score at baseline (SD) | CS use at baseline (%) | NSIST use at baseline (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ADAPT | Efgartigimod | 47.0 (14.7)† | 70.5† | 84.5† | NR | AChR+: 77%†,‡ MuSK+: 4%† Seronegative: 19%† | 9.5 (8.3)† “Time since gMG diagnosis” | II: 38.9%† III: 57.5%† IV: 3.6%† | 9.0 (2.5)† | 15.9 (4.8)† | 76† “Any steroid as MG therapy at baseline” | 61† “Any NSIST as MG therapy at baseline” | [10] |
| ADAPT-SC | Efgartigimod IV vs efgartigimod SC | 53.5§ | 59.1 | 91.8 | NR | NR | NR | NR | NR | NR | NR | NR | [44] |
| AGRX-113-1602 | Efgartigimod | 49.4 (17.4) | 62.5 | 91.7 | NR | AChR+: 100% | 10.8 (10.3) “MG duration” | II: 54.2% III: 41.7% IV: 4.2% | 8.0 (2.6) | 13.2 (5.9) | 54.2 “SOC: CS” | 50.0 “SOC: IS” | [45] |
| MycarinG | Rozanolixizumab | 51.8 (16.3) | 61 | 68 | NA: 30% EU: 60% Asia: 10% | AChR+: 90% MuSK+: 11% | 5.8 (2.5–10.6)¶ “Duration of disease” | II: 39% III: 57% IV: 4% | 8.3 (3.4) | 15.6 (3.6) | 65 “Medications: CS for systemic use” | 52 “Medications: IS” | [11] |
| Bril (2021) | Rozanolixizumab | 51.9 (15.1) | 62.8 | NR | NA: 20.9% EU: NR Asia: NR SA: NR | AChR+: 93% MuSK+: 2% | NR | II: 44.1%† III: 48.9%† IV: 7.0%† | 7.1 (3.0)† | 15.7 (3.9)† | 47 “MG treatments at baseline: CS (for systemic use)” | 49 “MG treatments at baseline: IS” | [46] |
| Vivacity-MG3 | Nipocalimab | 52.4 (15.97) | 60.1 | 62.7 | US: 12% East Asia: 31% EU/ROW: 57% | AChR+: 87.6% MuSK+: 10.5% LRP4+: 2.0% Seronegative: 0% | 7.9 (7.83) “Duration of MG” | II: 24.8%† III: 62.1%† IV: 12.4%† | 9.2 (2.38) | 15.4 (4.85) | 66.0† “Stable gMG therapy: CS for systemic use” | 53.6† “Stable gMG therapy: IS” | [12] |
| REGAIN | Eculizumab | 47.2 (16.8) | 66 | 76 | NA: 36.8%† EU: 40.8%† Asia: 12.8%† SA: 9.6%† | AChR+: 100% | 9.6 (8.2) “MG duration” | II: 37.6% III: 52.8% IV: 9.6% | 10.2 (2.8) | 17.1 (5.3) | 78 “CS at baseline” | NR | [13] |
| CHAMPION-MG | Ravulizumab | 55.6 (15.1) | 51.0 | 73 | NA: 45.7%† EU: 36.6%† Asia: 17.7%† | AChR+: 100% | 9.9 (9.3) “Time since gMG diagnosis” | II: 44.6% III: 49.1% IV: 7.4% | 9.0 (2.5) | 14.7 (5.2) | 69 “Use of GC at baseline” | 68 “Use of other IST# at baseline” | [14] |
| RAISE | Zilucoplan | 53.0 (15.2)† | 56.5† | 73.5† | NA: 52.2%† EU: 38.5%† Asia: 9.2%† | AChR+: 100% | 9.1 (10.0)† “Duration of disease” | II: 28.1% III: 67.2% IV: 4.6% | 10.6 (3.0)† | 19.1 (4.1)† | 63† “Baseline MG medications: CS” | NR | [15] |
| Howard (2020) | Zilucoplan | 49.4 (15.6)† | 52.3† | 81.8† | NR | AChR+: 100% | ZIL 0.1 mg: 6.5 (1.6–24.1)†† ZIL 0.3 mg: 5.3 (0.5–26.0)†† PBO: 6.3 (0.1–20.9)†† | II: 38.6%† III: 52.3%† IV: 9.1%† | 7.8 (3.2)† | 18.8 (4.4)† | 70.5† “MG therapy at baseline: prednisone” | NR | [47] |
| RINOMAX | Rituximab | 63.0 (16.0)† | 29.8† | NR | EU: 100% | AChR+: 95.7%† | 0.38 (0.25)†,‡‡ “Time since onset of gMG” | II: 44.1%† III: 55.9%† IV: NR | 4.8 (3.0)† | 9.4 (4.4)† | 59.6† “Prednisolone at baseline” | 0 (Per exclusion criteria) | [48] |
| BeatMG | Rituximab | 55.1 (17.1) | 44.2 | NR | NA: 100% | AChR+: 100% | 5.5 (5.9) “Time from diagnosis to randomization” | II: 59.6% III: 34.6% IV: 3.9% | 4.9 (3.6) | 10.1 (4.5) | 100 (Per inclusion criteria) | 34.6 “Baseline therapy: prednisone + IST” | [49] |
| NCT02473952 | Non-acute IVIg | 51.2 (15.63) | 53.2 | 95.2 | NA: 37.1%† EU: 62.9%† | AChR+: 100% | NR | NR | NR | NR | NR | NR | [50] |
| Bril (2023) | Non-acute IVIg | 48.1 (15.7) | 56.7 | 90.0 | NA: 40% EU: 60% | AChR+: 100% | 8.17 (6.91) “Time since MG diagnosis” | NR | 5.2 (4.0) | 11.6 (6.7) | 100 (Per inclusion criteria) | 58.3 “Other NSIST§§” | [51] |
| Wolfe (2002) | Non-acute IVIg | 41.1† (NR) | NR | NR | US: 100% | AChR+: 100% | NR | NR | 5.7 (3.8)† | 10.2 (4.5)† | 53† | NR | [39] |
†
Calculated using arm-level data.
‡
Reported outcome data for AChR+ patients.
§
Median since mean not reported.
¶
Median (IQR) since mean (SD) not reported.
#
Other ISTs included azathioprine, ciclosporin, methotrexate, mycophenolate mofetil, and tacrolimus.
††
Reported as “duration of disease” and as median (range) since mean (SD) not reported.
‡‡
Converted from days to years (1 year = 365.25 days).
§§
NSIST that were part of the background regimen included azathioprine, mycophenolate mofetil, cyclosporine, methotrexate, and cyclophosphamide.
AChR+: Anti-acetylcholine receptor antibody-positive; CS: Corticosteroid; EU: Europe; gMG: Generalized myasthenia gravis; IQR: Interquartile range; IS: Immunosuppressant; IST: Immunosuppressive therapy; IVIg: Intravenous immunoglobulin; LRP4+: Anti-low-density lipoprotein receptor-related protein 4 antibody-positive; MG: Myasthenia gravis; mg: Milligram; MG-ADL: Myasthenia Gravis Activities of Daily Living; MGFA: Myasthenia Gravis Foundation of America; MuSK+: Anti-muscle-specific kinase antibody-positive; NA: North America; NCT: National Clinical Trial; NR: Not reported; NSIST: Nonsteroidal immunosuppressant therapy; QMG: Quantitative Myasthenia Gravis; ROW: Rest of world; SA: South America; SD: Standard deviation; SOC: Standard of care; ZIL: Zilucoplan.
| Trial name | Intervention | MG-ADL CFB | MG-ADL response | QMG CFB | QMG response |
|---|---|---|---|---|---|
| ADAPT† | Efgartigimod | Reported for week 1–8 [4], 10 | Reported for MPI of 2-9 (week 4) | Reported for week 1–8 [4], 10 | Reported for MPI of 3-10 (week 4) |
| ADAPT-SC | Efgartigimod | Reported for week 1–8, 10 | Reported for MPI of 2 (week 10) | Reported for week 1–8, 10 | Reported for MPI of 3 (week 10) |
| ARGX-113-1602 | Efgartigimod | Reported for week 1–7 [5], 9, 11 (i.e., day 8, 15, 22, 29, 36, 43, 50, 64, 78) | Reported for MPI of 2-10 (week 4–5) | Reported for week 1–7 [5], 9, 11 (i.e., day 8, 15, 22, 29, 36, 43, 50, 64, 78) | NR |
| MycarinG | Rozanolixizumab | Reported for week 1-6 (i.e., day 8, 15, 22, 29, 36, 43) | Reported for MPI of 2-13 (week 6) | Reported for week 1-6 (i.e., day 8, 15, 22, 29, 36, 43) | Reported for MPI of 3-17 (week 6) |
| Bril (2021) | Rozanolixizumab | Reported for week 4 (i.e., day 29) | Reported for MPI of 2-10 (week 4) | Reported for week 0-4 (i.e., day 1, 8, 15, 22, 29) | Reported for MPI of 3-8 (week 4) |
| Vivacity-MG3 | Nipocalimab | Reported for week 2, 4, 6, 8, 10, 12, 14, 16, 18, 20, and 22/23/24 | Reported for MPI of 2-8 (week 24) | Reported for week 2, 4, 6, 8, 10, 12, 14, 16, 18, 20, and 22/24 | Reported for MPI of 3-9 (week 24) |
| REGAIN | Eculizumab | Reported for week 26 | Reported for MPI of 3-8 (week 26) | Reported for week 26 | Reported for MPI of 5-10 (week 26) |
| CHAMPION-MG | Ravulizumab | Reported for week 1, 2, 4, 10, 12, 18, 26 | Reported for MPI 2–6 [3] (week 26) | Reported for week 1, 2, 4, 10, 12, 18, 26 | Reported for MPI of 3–8 [5] (week 26) |
| RAISE | Zilucoplan | Reported for week 1, 2, 4, 8, 12 | Reported for MPI of 0–14 [3] (week 12) | Reported for week 1, 2, 4, 8, 12 | Reported for MPI of 0–18 [5] (week 12) |
| Howard (2020) | Zilucoplan | Reported for week 1, 2, 4, 8, 12 | NR | Reported for week 1, 2, 4, 8, 12 | Reported for MPI of 0–14 [3] (week 12) |
| RINOMAX | Rituximab | Reported for week 16, 24, 36, 48 | NR | Reported for week 16, 24, 36, 47 | NR |
| BeatMG | Rituximab | Reported for week 52 | NR | Reported for week 52 | Reported for MPI of 3-8 (week 52) |
| NCT02473952 | Non-acute IVIg | NR | NR | Reported for week 24 | NR |
| Bril (2023) | Non-acute IVIg | NR | NR | NR | NR |
| Wolfe (2002) | Non-acute IVIg | Reported for week 6 | NR | Reported for week 6 | NR |
†
For ADAPT, the principal timepoint of assessment for CFB outcomes was not clear and so was chosen based on the primary timepoint specified for response.
Bolded timepoints and MPIs are the principal ones reported for each trial.
CFB: Change from baseline; MG-ADL: Myasthenia Gravis Activities of Daily Living; MPI: Minimum point improvement; NR: Not reported; QMG: Quantitative Myasthenia Gravis.
Cross-trial differences in study design & treatment administration
Of the 15 RCTs, seven were phase ii trials and eight were phase III trials. Of the phase III trials, ADAPT (efgartigimod), CHAMPION-MG (ravulizumab), MycarinG (rozanolixizumab), RAISE (zilucoplan), REGAIN (eculizumab) and Vivacity-MG3 (nipocalimab) were registration trials. Whereas rozanolixizumab and zilucoplan were subcutaneous interventions, all others were administered intravenously. Dosing schedules and routes of administration varied across the included trials (Figure 2). Notably, in ADAPT, patients received four weekly infusions of efgartigimod per cycle, repeated based on waning MG-ADL response no sooner than 8 weeks after the start of the preceding cycle. In MycarinG, patients completed a 6-week treatment period with rozanolixizumab followed by an 8-week observation period, with outcomes measured throughout. The unique approaches employed in ADAPT and MycarinG pose challenges for conducting ITCs because patients in these trials improved while on treatment but worsened off-treatment, whereas in the trials that used maintenance dosing, patients experienced sustained improvement.

Figure 2. Dosing schedules (administration frequency and route) across randomized controlled trials included in the indirect treatment comparison feasibility assessment.
For the purposes of this figure, week 0 is the time of the first treatment dose.
ITC: Indirect treatment comparison; RCT: Randomized controlled trial.
The frequency of administration (maintenance dosing) also varied across trials, ranging from daily in the RAISE zilucoplan trial to every 8 weeks in the CHAMPION-MG ravulizumab trial, with most treatments being administered weekly or every 2 weeks (Figure 2).
Cross-trial differences in outcome definitions & assessment timepoints
All included RCTs reported CFB outcomes for MG-ADL and QMG. The principal timepoints at which these outcomes were assessed varied substantially across trials (Table 3), reflecting in part differences in study duration and dosing schedules. For example, the principal assessment timepoint for these outcomes ranged from 4 weeks in the ADAPT efgartigimod trial up to 52 weeks in the BeatMG rituximab trial.
The principal MPI values defining MG-ADL and QMG response also varied across trials (Table 3). Specifically, phase III trials for C5 complement inhibitors (eculizumab, ravulizumab and zilucoplan) used an MPI of 3 for MG-ADL and 5 for QMG, whereas other trials used MPIs of 2 and 3, respectively.
Cross-trial differences in patient populations
Trial eligibility criteria (e.g., Myasthenia Gravis Foundation of America [MGFA] class, antibody status, baseline MG-ADL score and treatment history) and patient characteristics at baseline (e.g., age, sex, race/ethnicity, geographical region, MGFA class, antibody status, disease duration, prestudy treatment exposure and disease severity) were generally well-reported across RCTs (Table 2 & Supplementary Table 3). Although assessed patient characteristics were broadly similar across trials, there were key differences in antibody status, time since gMG onset and prior treatment exposure.
Mean age at baseline and the proportion of female patients were reported for all included RCTs and were broadly comparable, with the exception of the RINOMAX rituximab trial population being relatively older and having a lower proportion of females (Table 2). Whereas trials of C5 complement inhibitors and non-acute IVIg consistently included only anti-AChR antibody-positive (AChR+) patients, trials for other treatments were less restrictive (Supplementary Table 3). The registration trials for FcRn inhibitors (ADAPT, MycarinG and Vivacity-MG3) were a mix of AChR+ and anti-AChR antibody-negative (AChR-) patients. However, where reported, data for the AChR+ subpopulation of these trials could be used to better align with other trials in ITC analyses.
The proportion of white patients, the proportion by geographical region, mean duration of disease, MGFA class at baseline, baseline MG-ADL score, baseline QMG score, the proportion receiving corticosteroids at baseline and the proportion receiving an NSIST at baseline were less well reported but broadly comparable across trials, with some exceptions (Table 2). Unlike other included trials, RINOMAX only included patients with new-onset gMG, as reflected by a lower baseline QMG score cutoff for inclusion, shorter average disease duration and lower baseline MG-ADL and QMG scores compared with other trials (Tables 1 & 2). Baseline MG-ADL and QMG scores were also relatively low for the BeatMG rituximab trial and the Bril et al. (2023) and Wolfe et al. (2002) non-acute IVIg trials. Unlike other included trials, the proportion receiving an NSIST at baseline was 0% in RINOMAX and the proportion receiving corticosteroids at baseline was 100% in BeatMG and the Bril et al. (2023) non-acute IVIg trial.
Cross-trial differences in treatment exposure were also noted. RINOMAX was the only trial to not permit prior thymectomy. The REGAIN eculizumab trial included relatively more treatment-refractory patients than the other trials (Table 1) wherein patients were required to have previously failed two or more immunosuppressive therapies or failed one or more immunosuppressive therapies with IVIg or plasma exchange given at least four-times per year for 12 months without symptom control.
Cross-trial differences in background treatments
Permitted and prohibited background treatments were broadly similar across included trials (Supplementary Table 4). Specifically, all trials permitted corticosteroids and NSISTs. However, the proportion of patients receiving specific background treatments, and their doses, routes and frequencies of administration, were not well reported across trials. Hence, while differences in background treatments may introduce bias in ITCs, it is difficult to assess the extent of the issue.
Feasibility assessment: assessment of network connectivity & placebo response
Prior to consideration of the comparability of placebo arms across included trials, a generalized connected network of treatments was visualized with placebo as a central node (Supplementary Figure 1). The network was star-shaped as all treatments (apart from the subcutaneous form of efgartigimod compared with intravenous efgartigimod in the ADAPT-SC trial) have been compared only with placebo. The network was also found to be sparse, as only one or two trials were available for each pair of connected treatments. Further, the timepoints at which MG-ADL and QMG outcomes were measured and the responder thresholds used differed across included trials (Table 3). These differences in outcomes are expected to impact the connectivity of networks.
While construction of a connected network anchored through placebo appears feasible, the comparability of placebo arms across trials is an important consideration in the conduct of anchored ITCs. For MG-ADL and QMG CFB, placebo response varied substantially across the included registration phase III RCTs (Supplementary Table 5). MG-ADL CFB for the placebo group ranged from -0.78 in the MycarinG rozanolixizumab trial (at week 6) to -3.25 in the nipocalimab Vivacity-MG3 trial (average of weeks 22, 23 and 24). Similarly, QMG CFB for the placebo group ranged from -0.8 in the CHAMPION-MG ravulizumab trial (at week 26) to -3.25 in the RAISE zilucoplan trial (at week 12). The differences in placebo response did not appear to be related to differences in reported trial characteristics, including dosing schedule or route of administration. For example, one might expect intravenous rather than subcutaneous dosing to be associated with a higher placebo response, but this was not consistently the case here. This suggests potential confounding by other sources of cross-trial heterogeneity, such as unmeasured or unreported patient baseline characteristics.
Feasibility assessment: identification of appropriate ITC methods
The assessments of trial characteristics and network connectivity identified several important considerations for selecting the most appropriate ITC analysis methods. There was evidence of within-trial imbalances in patient characteristics at baseline across included trials that could potentially bias NMA results. Although imbalances were present across all trials, they were more frequent in smaller non-registration trials. This finding suggests it may be appropriate to only consider large, well-balanced phase III registration trials in NMA base case analyses, while including the other trials in a sensitivity analysis to understand their impact on results. Still, the potential for residual confounding due to the observed imbalances in registration trials remains.
For included phase III registration RCTs, subgroup analyses of MG-ADL and QMG indicated the following characteristics may be TEMs: age, sex, race, geographical region, antibody status, disease duration, baseline MGFA class, baseline MG-ADL score and prior immunosuppressant therapy use (Supplementary Table 1). Several of these characteristics were noted to differ appreciably across included RCTs, which could introduce bias in ITCs due to violation of the exchangeability assumption. For example, the distribution of patients across relevant MG subgroups varied across trials, with some trials only including AChR+ patients. For trials including both AChR+ and AChR- patients, it may be appropriate to match the proportions of these groups in MAICs/STCs. In NMAs, it may be appropriate to restrict base case analyses to the AChR+ subpopulation to ensure comparability with trials of only AChR+ patients, with sensitivity analyses that include full trial populations to understand the impact of heterogeneity in serotypes across trials. As another example, the RINOMAX and REGAIN trial populations differed substantially from other trials with respect to potential TEMs (i.e., age, sex and baseline MG-ADL score for RINOMAX, and prior immunosuppressant therapy use for REGAIN). It may be appropriate to exclude RINOMAX and REGAIN as outlier trials from base case ITCs, with sensitivity analyses in which they are included.
An additional challenge in conducting anchored comparisons such as NMA is related to cross-trial differences in placebo arms (e.g., background treatments, administration route and schedule). Differences in placebo arm characteristics (as well as heterogeneity in measured and unmeasured TEMs) can result in systematic differences in placebo response, which can lead to bias in anchored ITC results. Indeed, substantial differences across the included trials in placebo response rates for MG-ADL and QMG were observed. Since the evidence network for gMG treatments is ‘star’ shaped with all RCTs but one linked via placebo, the validity of NMA is likely to be particularly dependent on the assumption that placebo arms across trials are comparable.
Given the within- and cross-trial variations in baseline characteristics, differences in the implementation of placebo arms, and the observed heterogeneity in placebo response rates across trials, there is a significant risk of bias in the results of anchored ITC approaches such as NMA. Although extensions of NMA such as baseline risk-adjusted NMA and network meta-regression can adjust for differences in TEMs across trials, they are not particularly useful and may not even be feasible in this situation given the sparseness of the evidence network (i.e., only one or two trials for each intervention). For the same reason, exclusion of outlier trials expected to bias ITC results will prevent some treatments (e.g., eculizumab) from being included in NMA, thereby potentially reducing the clinical relevance of ITCs.
Anchored MAICs/STCs, like NMAs, take advantage of the availability of a shared comparator between RCTs to compare relative outcomes in two treatment arms, thus maintaining randomization and the presumed balance of prognostic factors between trials. Unlike an NMA, an MAIC or STC leverages IPD to adjust for differences in reported patient characteristics (i.e., TEMs) that could otherwise bias ITC results. These methods were deemed feasible and are less likely to bias results compared with NMA. However, potential bias due to systematic cross-trial differences in placebo response discussed earlier, cannot be addressed by anchored MAICs/STCs. Similar to NMA, anchored MAICs may also give biased results in the presence of within-trial imbalances in patient characteristics. Arm-level (rather than trial-level) adjustments are a potential approach to address this concern, however they may break randomization, potentially introducing bias by distorting the balance between treatment arms of covariates not subjected to adjustment [52].
As an alternative, unanchored MAICs/STCs can help address the limitations associated with cross-trial placebo arm heterogeneity and imbalances within trials because they do not rely on the use of a shared comparator. Notably, unanchored analyses compare absolute outcomes of treatment arms from different trials, therefore differences in all prognostic factors and TEMs need to be adjusted for to avoid bias in the results [42]. Thus, unanchored adjusted ITC methods may be considered for treatment comparisons in gMG given the identified differences in placebo arm characteristics and within-trial imbalances.
Given the unique strengths and limitations of each ITC method in addressing the challenges posed by gMG trials, the use of multiple methods is warranted to provide a more holistic approach in assessing the comparative effectiveness of gMG treatments (Table 4, Figure 3). Conducting different ITC methods and comparing their results can help to clarify the extent to which methods may be biased. If all methods produce results in the same direction and of similar magnitude, this can increase confidence in the validity and reliability of comparative efficacy estimates. Alternatively, if there are differences in the results from each method, this can suggest further investigation is warranted to identify the most reliable method for the comparison(s) of interest.
| ITC method | Advantages | Disadvantages | ITC recommendations for gMG |
|---|---|---|---|
| NMA and Bucher ITC | • NMA permits multiple treatment comparisons within a single analysis • Methods are widely used and accepted by healthcare decision makers • Methods leverage randomization to account for cross-trial differences in patient characteristics that are PFs | • Ability to adjust for cross-trial differences in patient characteristics that are TEMs is limited, especially for sparse networks, therefore results may be prone to bias • Within-trial imbalances may lead to residual confounding of uncertain magnitude and direction • Methods are dependent on the availability of common comparators (e.g., anchoring treatments) that are assumed to be homogeneous, even if routes of administration differ | • Anchored MAICs/STCs can adjust for imbalanced TEMs • Unanchored MAICs/STCs can address the limitations of using placebo as a common comparator and residual confounding due to within-trial imbalances in patient characteristics • Both anchored and unanchored comparisons should be conducted, as anchored comparisons might underestimate or overestimate the true clinical benefit due to observed between-trial heterogeneity and connectivity challenges • Identification of TEMs and PFs in gMG can be challenging. A comprehensive approach leveraging a literature review, input from clinical experts, and analysis of trial subgroup data is recommended. • Conducting ITCs using outcome data for a single assessment timepoint may introduce bias (e.g., favoring one treatment over another based on the chosen timepoint); methods that incorporate multiple timepoints may be appropriate • Change from baseline outcomes may be more appropriate for ITCs than response outcomes • Non-registration trials often have smaller sample sizes and larger within-trial imbalances, so they should be included only as sensitivity analyses (i.e., base case analyses should not include rituximab or non-acute IVIg) • Critical differences in study and population characteristics including background treatments and treatment administration cannot always be adjusted for in ITCs • No single ITC method can be deemed most appropriate, so multiple methods should be employed, each accompanied by appropriate sensitivity analyses |
| Anchored MAIC/STC | • Methods can adjust for cross-trial differences in patient baseline characteristics that are TEMs • Methods leverage randomization to account for cross-trial differences in patient characteristics that are PFs | • Within-trial imbalances may lead to residual confounding of uncertain magnitude and direction • Methods are dependent on the availability of common comparators (e.g., anchoring treatments) that are assumed to be homogeneous, even if routes of administration differ • Methods assume all important TEMs are captured in reported trial data for treatments being compared • Comparisons with poor overlap will translate into lower ESS, resulting in increased uncertainty, such that results may not be considered sufficiently robust to inform decision-making • STCs are less widely used than MAICs and more challenging to communicate | |
| Unanchored MAIC/STC | • Methods do not require a connected network (i.e., an anchoring treatment) • Methods are not dependent on the availability of a common comparator across trials | • Methods require adjustment for PFs as well as TEMs • Methods assume all important TEMs and PFs are captured in reported trial data for treatments being compared • Comparisons with poor overlap have lower ESS, resulting in increased uncertainty, such that results may not be considered sufficiently robust to inform decision-making • STCs are less widely used than MAICs and more challenging to communicate |
ESS: Effective sample size; gMG: Generalized myasthenia gravis; ITC: Indirect treatment comparison; IVIg: Intravenous immunoglobulin; MAIC: Matching-adjusted indirect comparison; NMA: Network meta-analysis; PF: Prognostic factor; RCT: Randomized controlled trial; STC: Simulated treatment comparison; TEM: Treatment effect modifier.
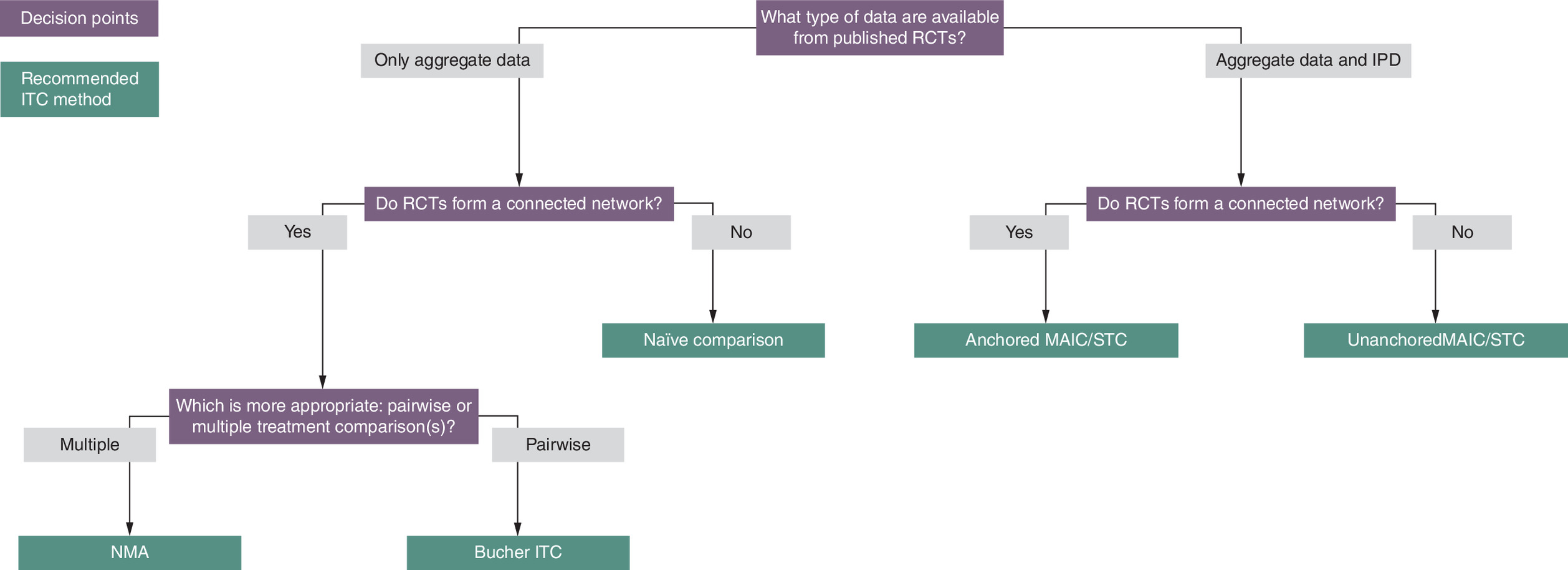
Figure 3. Indirect treatment comparison method selection algorithm.
Adapted from Tanaka et al. with permission [53].
IPD: Individual patient data; ITC: Indirect treatment comparison; MAIC: Matching-adjusted indirect comparison; RCT: Randomized controlled trial; STC: Simulated treatment comparison.
Differences in reported timepoints may impact the validity of ITC analyses. For any outcome measured over time, the choice of timepoint is critical to minimize bias from this factor in an ITC. Ideally the same timepoint should be used across trials, but this is difficult in practice given variation in follow-up times and the frequency of outcome assessment across RCTs. Even in situations where outcomes are reported for a comparable timepoint, fluctuations in treatment response due to cyclical dosing schedules may favour one treatment over another in an ITC. The timepoint(s) selected for analysis may reflect a period where treatment is or is not being received. For example, in the ADAPT trial, the primary assessment timepoint (week 4) reflected the end of the treatment cycle but does not account for potential waning of effect during off-treatment. Hence, regardless of the ITC method used, it may be valuable to conduct analyses using outcome data representative of multiple timepoints (e.g., taking the average across several timepoint or using an area under the curve [AUC] approach to compare the longer term efficacy of cyclical vs continuous treatments), as opposed to a single timepoint.
Finally, MG-ADL and QMG response definitions and assessment timepoints for the principal reported outcomes generally varied between RCTs. In particular, trials for C5 complement inhibitors used higher responder thresholds than trials for FcRn inhibitors. It is important to account for this difference when conducting ITCs, which could involve using other reported response data to achieve alignment in MPI where possible. Many of the included trials reported response outcomes for multiple MPI values. However, in cases where it is not possible to align on MPI values, ITCs for response outcomes with varying MPIs may still warrant investigation, so long as the limitations of such an approach are acknowledged.
Discussion
Summary of findings
In this study we performed a formal feasibility assessment to systematically evaluate currently available trials for gMG treatments with the goal of identifying the advantages and disadvantages of different ITC methods. The current study highlights the importance of conducting a thorough ITC feasibility assessment, as failing to do so may lead to use of ITC methods yielding biased results. Several barriers to conducting robust ITCs in gMG were identified, including within-trial imbalances in patient characteristics, small trial sizes and cross-trial differences in potential TEMs. Further, heterogeneity in placebo administration characteristics, background therapies and cross-trial variation in placebo response for key outcomes were noted. Treatment strategies (i.e., cyclical vs continuous), dosing schedules and outcome assessment timepoints were inconsistent across trials, necessitating careful consideration of methods and timepoints when interpreting outcomes. The use of multiple ITC methods, with careful evaluation of underlying assumptions and limitations, can help limit bias and provide a more complete understanding of the comparative effectiveness of gMG treatments.
We also identified the need for methodological approaches that address the heterogeneity in treatment strategy and outcome assessment timepoints across RCTs for gMG therapies. The issues of cyclical versus continuous treatment approaches and varying assessment timepoints are independent of the choice of ITC method (NMA, anchored/unanchored MAIC/STC) as none of these methods can adjust for such differences. As gMG is a chronic disease, timepoints reflecting longer term efficacy are generally of greatest interest to decision makers. However, assessment of long-term efficacy for cyclically-dosed treatments is challenging given that the reported data from trials reflect timepoints associated with maximal effects and/or waning of treatment benefit. The potential for dosing regimens of cyclical therapies in clinical practice to differ from dosing in trials should also be considered. Approaches employing average treatment effects or AUC may hold promise for analyzing data for cyclical therapies in ITCs; however, they do not capture patient or clinician preference for consistent versus fluctuating disease control.
Comparison with previous research
Several of the issues associated with conducting ITCs in gMG identified in our study have been reported in previous HTA agency reviews. Between-trial population differences and their implications for appropriate ITC methods identified in the current study were highlighted by Canada’s Drug Agency (CDA) and NICE in their reviews of efgartigimod and ravulizumab. The CDA efgartigimod review noted results of an NMA comparing efgartigimod, ravulizumab and non-acute IVIg were uncertain due to important differences between gMG trials (including variability in trial eligibility criteria) as well as inconsistent and insufficiently detailed reporting of baseline patient characteristics [22]. The ravulizumab submissions to NICE and CDA included Bucher ITC, MAIC and IPD-only analyses comparing ravulizumab with eculizumab [23,26]. Both HTA agencies noted differences in inclusion criteria related to prior therapy between REGAIN and CHAMPION-MG (i.e., REGAIN required patients to have failed on immunosuppressive drugs, while CHAMPION-MG did not). The critical appraisal by CDA mentioned a small effective sample size for submitted MAIC and IPD-only analyses as evidence of limited overlap between trial populations and concluded an unadjusted ITC between CHAMPION-MG and REGAIN was unlikely to be valid. In reviews of efgartigimod and ravulizumab, NICE noted the availability of two rituximab trials (BeatMG and RINOMAX) but did not push for their inclusion in ITCs due to their small sizes and lack of statistically significant results versus placebo [26,54]. Taken together, the NICE and CDA reviews supported our identification of REGAIN and RINOMAX as potential outlier trials to consider excluding from base case analyses and more generally the importance of conducting adjusted ITCs to account for population differences where possible.
The CDA and NICE ravulizumab reviews also identified the importance of clear and detailed reporting of ITC analyses, including justifications for the ITC methods selected, how trials and outcomes to be compared are identified, and the selection process by which covariates to be included in adjusted analyses are identified [23,26]. Specifically, NICE raised concerns about a less-than-comprehensive set of covariates having been used in adjusted ITC analyses, as well a lack of sensitivity analyses to evaluate the robustness of analyses with adjustments for different sets of covariates. A comprehensive and transparent approach to identifying and justifying covariates for adjustment in MAICs/STCs is critical to optimize the adjustment process and thereby minimize bias introduced by cross-trial differences in patient populations. In a review of published approaches to selecting effect modifiers for adjustment in ITCs, Freitag et al. recommended a three-pronged approach for identifying TEMs: systematic reviews; expert input and assessment of the distribution of TEMs (e.g., examining subgroup analyses in clinical trials) [55].
Aligned with our findings, challenges for ITC analyses due to cross-trial variation in dosing schedules, follow-up times and primary assessment timepoints for MG-ADL and QMG outcomes were noted by both CDA and NICE in their reviews of gMG treatments, as well as in an EMA review of zilucoplan [22–24,26,54]. Specifically, the CDA review of efgartigimod highlighted its unique dosing schedule (i.e., symptom-based cyclical dosing, as opposed to continuous dosing for comparators) and the range of study follow-up times as important differences between trials for gMG treatments that increase uncertainty and limit the interpretability of ITC results [22]. We also noted using the primary assessment timepoint from each trial in an ITC could introduce bias for or against some treatments, whereas conducting ITCs at an early timepoint (e.g., week 4) could bias against treatments demonstrating improved response over time. In their ITC study, Fawsitt et al. discussed the importance of considering different analytical approaches where there are cross-study differences in the timepoints for which CFB outcome data are available, and specifically used linear interpolation to estimate treatment effects at timepoints not reported in comparator studies [35]. In their publication describing a process for assessing NMA feasibility, Cope et al. suggested considering ITC approaches that combine multiple timepoints for outcomes measured over time [37]. Considering only a single timepoint may be insufficient to capture variations in response resulting from varying dosing schedules. Analyzing data at a single timepoint can introduce bias, favoring one treatment over another based on the chosen timepoint. As with ITC methods more broadly, it may be appropriate to conduct ITC analyses for gMG treatments using different timepoint data for CFB outcomes: using a single reported timepoint (e.g., per protocol definition), taking the average across multiple timepoints, or calculating the AUC. For example, an EMA report on zilucoplan included submitted ITCs using AUC for MG-ADL and QMG CFB outcomes, which were described as measuring an aggregated effect over time to facilitate comparison between efgartigimod (with cyclical dosing and effect in ADAPT) and zilucoplan (with continuous dosing and a sustained effect in RAISE) [24]. There is a need for further investigation of the strengths and weaknesses of these approaches for ITCs of gMG treatments, but ultimately it is important to explore multiple approaches to provide a comprehensive view of how differences in timepoints can impact ITC results.
Supporting our finding of substantial cross-trial variation in MG-ADL and QMG placebo response, a relatively high placebo response was also recently reported for the LUMINESCE phase III satralizumab trial and the MINT phase III inebilizumab trial, which were published after the search dates for our SLR [56,57]. Placebo arm heterogeneity and its impact on ITC results has been the focus of previous research with potential implications for ITCs of gMG treatments. Specifically, recent ITC publications have identified unanchored ITCs as appropriate approaches where there is evidence of placebo arm heterogeneity [35,58,59]. Prior to conducting ITCs for treatments for postpartum depression. Meltzer-Brody et al. evaluated the placebo arms of included studies [58]. The authors noted the placebo arm of one trial had a substantially larger CFB for outcomes of interest, which they attributed to more frequent in-person study visits or expectations regarding the therapy [58]. Because of this difference, they determined the placebo arm was not an appropriate common comparator in ITCs and opted to conduct unanchored MAICs [58]. To explore how differences in route of administration affect the results of ITCs where placebo is the common comparator, Fawsitt et al. compared anchored and unanchored ITC methods using CFB outcome data from placebo-controlled RCTs of subcutaneous and intravenous migraine preventative treatments [35]. The authors found results varied based on the ITC method used and concluded that in situations where there is cross-trial heterogeneity in placebo characteristics, decision makers should consider conducting unanchored ITCs in addition to NMA [35]. Fawsitt et al. further noted differences in routes of administration may influence patient perceptions regarding treatment efficacy and thereby affect the size of placebo response for patient-reported outcomes [35]. As such, anchored ITCs based on evidence networks of treatments with different routes of administration may be susceptible to expectation bias [35].
Several ITCs of gMG treatments have recently been published [60–63]. An examination of these publications (e.g., reported limitations) can help inform future ITCs in this therapeutic area. Ma et al. and Chen et al. each conducted NMAs using phase II and III RCTs for both approved and unapproved gMG treatments [60,61]. The authors of both studies acknowledged the small sample sizes of some included trials and cross-trial patient population differences as limitations of their analyses. A commentary on Chen et al. raised concerns regarding their target population not being clearly defined with respect to whether it included AChR+ and/or MuSK+ patients and criticized the lack of discussion around cross-trial differences in TEMs and outcome assessment timepoints [27]. Aligned with our findings, Chen et al. identified rituximab trials as being associated with higher heterogeneity and removed RINOMAX in a sensitivity analysis [60]. Saccà et al. (2023) also recently conducted an exploratory NMA as part of a broader SLR and meta-analysis [62]. The authors noted differences in patient characteristics between trials for rituximab and other treatments but did not investigate the sources of heterogeneity. Other than rituximab, the authors included only registration phase III RCTs without explanation for how trials or treatments were selected. Two recent anchored MAIC analyses compared efgartigimod with ravulizumab and rituximab on MG-ADL CFB. However, neither reported a formal approach to identifying important TEMs and both considered only a single timepoint for analysis [64,65].
Strengths & limitations
Strengths of this study included the use of a systematic review to build an evidence base for the ITC feasibility assessment, the comprehensive assessment of trial characteristics in alignment with best practices, and thorough consideration of the advantages and disadvantages of multiple ITC methods in relation to the challenges posed by the gMG evidence base.
There were also certain limitations with the current study. First, we focused on MG-ADL and QMG outcomes, although other efficacy and safety outcomes are reported for RCTs in gMG. MG-ADL and QMG were considered to be the most relevant because they were the most commonly reported efficacy outcomes in the included RCTs, and further, were often the primary or key secondary outcomes in these trials. They are also widely accepted as the most relevant parameters for MG severity. Second, we identified potential within-trial imbalances and potential TEMs using semi-quantitative approaches that were not validated. Although we evaluated reported RCT subgroup analyses to identify potential TEMs, these varied across trials and so there is uncertainty as to what characteristics are the most important TEMs. Third, some characteristics were not consistently reported across trials, limiting our ability to characterize clinically important heterogeneity and potential impacts on the choice of ITC methods.
Conclusion
Across RCTs for add-on therapies in gMG, important differences were identified in potential TEMs, placebo characteristics (including background treatments, route of administration and response), treatment administration (cyclical vs continuous dosing, and varying dosing schedules), assessment timepoints and response definitions. Within-trial patient differences and small sample sizes were also noted. Collectively, these findings suggest use of anchored ITC approaches such as NMA and anchored MAIC/STC to compare treatments for gMG could generate biased results. Unanchored MAIC/STC have the advantage that a common comparator is not required, although the validity of these methods rests on more stringent requirements for adjustment of prognostic factors and TEMs. Regardless of ITC method, challenges associated with cyclical versus continuous dosing, varying timepoints of assessment and differences in response definition remain. These require careful methodological consideration to ensure ITC results reflect fair, clinically relevant comparisons between treatments. Given the different strengths and limitations of each ITC method, we conclude that multiple ITC methods should be used with careful evaluation of their underlying assumptions, and a comprehensive set of sensitivity analyses should be performed to ensure appropriate exploration of the breadth of heterogeneity observed across gMG trials. Consideration should be given to prioritizing the use of data from balanced trials of sufficiently large size (i.e., phase III RCTs) in ITCs. Finally, robust methods for identification of important covariates are of paramount importance to ensure the validity of population-adjusted ITCs.
Summary points
•
There are currently no head-to-head randomized controlled trials for immunotherapies added to standard of care in generalized myasthenia gravis (gMG), a rare autoimmune disorder involving disruption of neuromuscular junction signaling.
•
In the absence of direct evidence, indirect treatment comparisons (ITCs) are potentially valuable to understand the relative effectiveness of gMG therapies.
•
Multiple ITC methods are available and have different requirements, so a comprehensive assessment of available trials is important to understand the most appropriate approach to using ITCs to generate reliable comparative estimates.
•
A systematic literature review was conducted and identified 15 relevant clinical trials, which were then assessed by comparing their design, population and outcome characteristics.
•
Several barriers to conducting robust ITCs were identified, including within-trial imbalances in patient characteristics, small trial sizes and cross-trial differences in potential treatment effect modifiers.
•
Heterogeneity in placebo administration characteristics and background therapies, and cross-trial variation in placebo response for key outcomes were also noted.
•
Treatment strategies, dosing schedules and outcome assessment timepoints were inconsistent across trials, necessitating careful consideration when interpreting outcomes.
•
Multiple approaches to ITCs, with careful evaluation of underlying assumptions and limitations, are required to limit bias and ensure robust comparative efficacy estimates are available to decision makers.
Author contributions
All authors contributed to study conception and design. Data extraction and analysis were performed by A Nero and C Drudge. C Drudge wrote the first draft of the manuscript. All authors reviewed and revised the manuscript. All authors take responsibility for the integrity of the study and have approved the final version.
Acknowledgments
The authors thank V Barabash, B Salvo and J Ellis, who contributed to this work as part of their roles as paid employees of EVERSANA.
Financial disclosure
This work was funded by Johnson & Johnson, NJ, USA. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Competing interests disclosure
NE Gilhus has received consultative or speaker’s honoraria from Johnson & Johnson, UCB, Argenx, Alexion, Merck, Dianthus, Amgen, Roche, Grifols, Immunovant, Huma, Denka and Takeda. S Jacob has served as an international advisory board member or has been in the data monitoring committee for clinical trials for Alexion, Alnylam, Argenx, Johnson & Johnson, Immunovant, Merck, Novartis, Regeneron and UCB pharmaceuticals, is currently an expert panel member of Myasthenia Gravis consortium for Argenx pharmaceuticals and has received speaker fees from Argenx, Eisai, Terumo BCT and UCB pharmaceuticals. He is also a board member (trustee) of the UK myasthenia patient charity, Myaware. M Hashim, SV Sanden and K Gandhi are employees of Johnson & Johnson and may hold stock/stock options of Johnson & Johnson. C Drudge, A Nero and S Singh are employees of EVERSANA. EVERSANA receives consultancy fees from pharmaceutical and device companies, including Johnson & Johnson. B Hutton has previously received honoraria from EVERSANA and Evidinno Outcomes Research Inc. for provision of methodologic advice related to the conduct of systematic reviews, meta-analyses and ITCs. The authors have no other competing interests or relevant affiliations with any organization or entity with the subject matter or materials discussed in the manuscript apart from those disclosed.
Writing disclosure
No funded writing assistance was utilized in the production of this manuscript.
Open access
This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 Unported License. To view a copy of this license, visit https://creativecommons.org/licenses/by-nc-nd/4.0/
Supplementary Material
File (supplementary material.docx)
- Download
- 615.67 KB
References
Papers of special note have been highlighted as: • of interest
1.
Gelinas D, Parvin-Nejad S, Phillips G et al. The humanistic burden of myasthenia gravis: a systematic literature review. J. Neurol. Sci. 437, 120268 (2022).
2.
Engebretsen I, Gilhus NE, Kristiansen IS et al. The epidemiology and societal costs of myasthenia gravis in Norway: a non-interventional study using national registry data. Eur. J. Neurol. 31(5), e16233 (2024).
3.
Saccà F, Salort-Campana E, Jacob S, Cortés-Vicente E, Schneider-Gold C. Refocusing generalized myasthenia gravis: patient burden, disease profiles, and the role of evolving therapy. Eur. J. Neurol. 31(6), e16180 (2024).
4.
Zhdanava M, Pesa J, Boonmak P et al. Economic burden of generalized myasthenia gravis (MG) in the United States and the impact of common comorbidities and acute MG-related events. Curr. Med. Res. Opin. 40(7), 1145–1153 (2024).
5.
Gilhus NE, Tzartos S, Evoli A, Palace J, Burns TM, Verschuuren JJGM. Myasthenia gravis. Nat. Rev. Dis. Primers. 5(1), 30 (2019).
6.
Sanders DB, Wolfe GI, Benatar M et al. International consensus guidance for management of myasthenia gravis. Neurology 87(4), 419–425 (2016).
7.
Nair SS, Jacob S. Novel immunotherapies for myasthenia gravis. Immunotargets Ther. 12, 25–45 (2023).
8.
Narayanaswami P, Sanders DB, Wolfe G et al. International consensus guidance for management of myasthenia gravis. Neurology 96(3), 114–122 (2021).
9.
Wiendl H, Abicht A, Chan A et al. Guideline for the management of myasthenic syndromes. Ther. Adv. Neurol. Disord. 16, 17562864231213240 (2023).
10.
Howard JF Jr, Bril V, Vu T et al. Safety, efficacy, and tolerability of efgartigimod in patients with generalised myasthenia gravis (ADAPT): a multicentre, randomised, placebo-controlled, Phase III trial. Lancet Neurol. 20(7), 526–536 (2021).
11.
Bril V, Drużdż A, Grosskreutz J et al. Safety and efficacy of rozanolixizumab in patients with generalised myasthenia gravis (MycarinG): a randomised, double-blind, placebo-controlled, adaptive Phase III study. Lancet Neurol. 22(5), 383–394 (2023).
12.
Antozzi C, Vu T, Ramchandren S et al. Safety and efficacy of nipocalimab in adults with generalised myasthenia gravis (Vivacity-MG3): a Phase III, randomised, double-blind, placebo-controlled study. Lancet Neurol. 24(2), 105–116 (2025).
13.
Howard JF Jr, Utsugisawa K, Benatar M et al. Safety and efficacy of eculizumab in anti-acetylcholine receptor antibody-positive refractory generalised myasthenia gravis (REGAIN): a Phase III, randomised, double-blind, placebo-controlled, multicentre study. Lancet Neurol. 16(12), 976–986 (2017).
14.
Vu T, Meisel A, Mantegazza R et al. Terminal complement inhibitor ravulizumab in generalized myasthenia gravis. NEJM Evid. 1(5), EVIDoa2100066 (2022).
15.
Howard JF Jr, Bresch S, Genge A et al. Safety and efficacy of zilucoplan in patients with generalised myasthenia gravis (RAISE): a randomised, double-blind, placebo-controlled, Phase III study. Lancet Neurol. 22(5), 395–406 (2023).
16.
Salanti G. Indirect and mixed-treatment comparison, network, or multiple-treatments meta-analysis: many names, many benefits, many concerns for the next generation evidence synthesis tool. Res. Synth. Methods 3(2), 80–97 (2012).
• Describes network meta-analysis (NMAs), which are one of the indirect treatment comparison (ITC) methods considered in this article.
17.
Signorovitch JE, Sikirica V, Erder MH et al. Matching-adjusted indirect comparisons: a new tool for timely comparative effectiveness research. Value Health 15(6), 940–947 (2012).
• Describes matching-adjusted indirect comparisons (MAICs), which are one of the ITC methods considered in this article.
18.
Lu G, Ades AE. Combination of direct and indirect evidence in mixed treatment comparisons. Stat. Med. 23(20), 3105–3124 (2004).
19.
Bucher HC, Guyatt GH, Griffith LE, Walter SD. The results of direct and indirect treatment comparisons in meta-analysis of randomized controlled trials. J. Clin. Epidemiol. 50(6), 683–691 (1997).
20.
Ishak KJ, Proskorovsky I, Benedict A. Simulation and matching-based approaches for indirect comparison of treatments. Pharmacoeconomics 33(6), 537–549 (2015).
21.
European Union. HTA CG Methodological Guideline for Quantitative Evidence Synthesis: Direct and Indirect Comparisons. (2024). Available from: https://health.ec.europa.eu/publications/methodological-guideline-quantitative-evidence-synthesis-direct-and-indirect-comparisons_en
• Describes available ITC methods and provides general guidance on which methods are appropriate for particular situations.
22.
Canada's Drug Agency. Reimbursement recommendation for efgartigimod alfa (Vyvgart). (2023). Available from: https://www.cadth.ca/sites/default/files/DRR/2024/SR0782REC-Vyvgart-meta.pdf
23.
Canada's Drug Agency. Reimbursement recommendation for ravulizumab (Ultomiris). (2023). Available from: https://www.cda-amc.ca/sites/default/files/DRR/2023/SR0765Ultomiris%20-%20Confidential%20Final%20CADTH%20Recommendation%20August%2024%2C%202023%20revised.pdf
24.
European Medicines Agency. Orphan designation withdrawal assessment report for Zilbrysq (zilucoplan). (2023). Available from: https://www.ema.europa.eu/en/documents/orphan-maintenance-report/zilbrysq-orphan-designation-withdrawal-assessment-report_en.pdf
25.
Institute for Clinical and Economic Review. Eculizumab and efgartigimod for the treatment of myasthenia gravis: effectiveness and value. Final Evid.Rep. (2021). Available from: https://icer.org/assessment/myasthenia-gravis/
26.
National Institute for Health and Care Excellence. Ravulizumab for treating generalised myasthenia gravis (terminated appraisal). Technology appraisal TA940. Com.Pap. (2023). Available from: https://www.nice.org.uk/guidance/ta940/documents/committee-papers
27.
Zhang I, Jansen JP, Yungher BJ, Kielhorn A, Yee KS. Commentary: efficacy and safety of the innovative monoclonal antibodies in adults with generalized myasthenia gravis: a Bayesian network analysis. Front. Immunol. 15, 1403802 (2024).
28.
Ades AE, Caldwell DM, Reken S, Welton NJ, Sutton AJ, Dias S. NICE DSU Technical Support Document 7: evidence synthesis of treatment efficacy in decision making: a reviewer's checklist. (2012). Available from: https://www.sheffield.ac.uk/nice-dsu/tsds/full-list
29.
Dias S, Sutton AJ, Welton NJ, Ades AE. NICE DSU Technical Support Document 3: heterogeneity: subgroups, meta-regression, bias and bias-adjustment. (2012). Available from: https://www.sheffield.ac.uk/nice-dsu/tsds/full-list
30.
Higgins Jpt TJ, Chandler J, Cumpston M, Li T, Page MJ, Welch VA (Eds). Cochrane Handbook for Systematic Reviews of Interventions version 6.4 (updated August 2023). Cochrane (2023). Available from: http://www.training.cochrane.org/handbook
• Provides detailed information on systematic literature reviews and NMAs, including important considerations when conducting these activities.
31.
Hoaglin DC, Hawkins N, Jansen JP et al. Conducting indirect-treatment-comparison and network-meta-analysis studies: Report of the ISPOR Task Force on Indirect Treatment Comparisons Good Research Practices: Part 2. Value Health 14(4), 429–437 (2011).
• Provides guidance on conducting ITCs including NMAs.
32.
Cameron C, Hutton B, Druchok C et al. Importance of assessing and adjusting for cross-study heterogeneity in network meta-analysis: a case study of psoriasis. J. Comp. Eff. Res. 7(11), 1037–1051 (2018).
33.
Nikolakopoulou A, Chaimani A, Furukawa TA, Papakonstantinou T, Rücker G, Schwarzer G. When does the placebo effect have an impact on network meta-analysis results? BMJ Evid. Based Med. 29(2), 127 (2024).
34.
Bannuru RR, Mcalindon TE, Sullivan MC, Wong JB, Kent DM, Schmid CH. Effectiveness and implications of alternative placebo treatments. Ann. Intern. Med. 163(5), 365–372 (2015).
35.
Fawsitt CG, Thom H, Regnier Stephane A, Lee Xin Y, Kymes S, Vase L. Comparison of indirect treatment methods in migraine prevention to address differences in mode of administration. J. Comp. Eff. Res. 12(7), e230021 (2023).
36.
Swerts DB, Benedetti F, Peres MFP. Different routes of administration in chronic migraine prevention lead to different placebo responses: a meta-analysis. Pain 163(3), 415–424 (2022).
37.
Cope S, Zhang J, Saletan S, Smiechowski B, Jansen JP, Schmid P. A process for assessing the feasibility of a network meta-analysis: a case study of everolimus in combination with hormonal therapy versus chemotherapy for advanced breast cancer. BMC Med. 12(1), 93 (2014).
• Outlines a process for assessing the feasibility of NMAs.
38.
Page MJ, Mckenzie JE, Bossuyt PM et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. Brit. Med. J. 372, n71 (2021).
39.
Wolfe GI, Barohn RJ, Foster BM et al. Randomized, controlled trial of intravenous immunoglobulin in myasthenia gravis. Muscle Nerve 26(4), 549–552 (2002).
40.
European Union. HTA CG Practical Guideline for Quantitative Evidence Synthesis: Direct and Indirect Comparisons. (2024). Available from: https://health.ec.europa.eu/publications/practical-guideline-quantitative-evidence-synthesis-direct-and-indirect-comparisons_en
• Describes available ITC methods and provides practical guidance on which methods are appropriate for particular situations.
41.
Jansen JP, Fleurence R, Devine B et al. Interpreting indirect treatment comparisons and network meta-analysis for health-care decision making: Report of the ISPOR Task Force on Indirect Treatment Comparisons Good Research Practices: Part 1. Value Health 14(4), 417–428 (2011).
42.
Phillippo DM, Ades AE, Dias S, Palmer S, Abrams KR, Welton NJ. NICE DSU Technical Support Document 18: methods for population-adjusted indirect comparisons in submission to NICE. (2016). Available from: https://www.sheffield.ac.uk/nice-dsu/tsds/full-list
43.
Austin PC. Balance diagnostics for comparing the distribution of baseline covariates between treatment groups in propensity-score matched samples. Stat. Med. 28(25), 3083–3107 (2009).
44.
ClinicalTrials.gov. Evaluating the pharmacodynamic noninferiority of efgartigimod PH20 SC administered subcutaneously as compared to efgartigimod administered intravenously in patients with generalized myasthenia gravis (ADAPT-SC). (2023). Available from: https://clinicaltrials.gov/study/NCT04735432
45.
Howard JF, Bril V, Burns TM et al. Randomized Phase II study of FcRn antagonist efgartigimod in generalized myasthenia gravis. Neurology 92(23), e2661–e2673 (2019).
46.
Bril V, Benatar M, Andersen H et al. Efficacy and safety of rozanolixizumab in moderate to severe generalized myasthenia gravis. Neurology 96(6), e853–e865 (2021).
47.
Howard JF Jr, Nowak RJ, Wolfe GI et al. Clinical effects of the self-administered subcutaneous complement inhibitor zilucoplan in patients with moderate to severe generalized myasthenia gravis: results of a Phase II randomized, double-blind, placebo-controlled, multicenter clinical trial. JAMA Neurol. 77(5), 582–592 (2020).
48.
Piehl F, Eriksson-Dufva A, Budzianowska A et al. Efficacy and safety of rituximab for new-onset generalized myasthenia gravis: the RINOMAX randomized clinical trial. JAMA Neurol. 79(11), 1105–1112 (2022).
49.
Nowak RJ, Coffey CS, Goldstein JM et al. Phase II trial of rituximab in acetylcholine receptor antibody-positive generalized myasthenia gravis. Neurology 98(4), e376–e389 (2022).
50.
ClinicalTrials.gov. A study to evaluate the efficacy and safety of IGIV-C in symptomatic subjects with generalized myasthenia gravis. (2019). Available from: https://clinicaltrials.gov/study/NCT02473952
51.
Bril V, Szczudlik A, Vaitkus A et al. Randomized double-blind placebo-controlled trial of the corticosteroid-sparing effects of immunoglobulin in myasthenia gravis. Neurology 100(7), e671–e682 (2023).
52.
Remiro-Azocar A, Heath A, Baio G. Methods for population adjustment with limited access to individual patient data: a review and simulation study. Res. Synth. Methods 12(6), 750–775 (2021).
53.
Tanaka S, Igarashi A, De Moor R et al. A targeted review of worldwide indirect treatment comparison (ITC) guidelines and best practices. Value Health 27(9), 1179–1190 (2024).
54.
National Institute for Health and Care Excellence. Efgartigimod for treating generalised myasthenia gravis (in development). Technology appraisal ID4003. Com.Pap. (2023). Available from: https://www.nice.org.uk/guidance/gid-ta10986/documents/committee-papers
55.
Freitag A, Gurskyte L, Sarri G. Increasing transparency in indirect treatment comparisons: is selectingeffect modifiers the missing part of the puzzle? A review of methodological approaches and critical considerations. J. Comp. Eff. Res. 12(10), e230046 (2023).
56.
Habib AA, Zhao C, Aban I et al. Safety and efficacy of satralizumab in patients with generalised myasthenia gravis (LUMINESCE): a randomised, double-blind, multicentre, placebo-controlled Phase III trial. Lancet Neurol. 24(2), 117–127 (2025).
57.
Amgen. Amgen presents positive Phase IIIdata for Uplizna (inebilizumab-cdon) in generalized myasthenia gravis (gMG) at AANEM 2024. (2024). Available from: https://www.amgen.com/newsroom/press-releases/2024/10/amgen-presents-positive-phase-3-data-for-uplizna-inebilizumabcdon-in-generalized-myasthenia-gravis-gmg-at-aanem-2024
58.
Meltzer-Brody S, Gerbasi ME, Mak C et al. Indirect comparisons of relative efficacy estimates of zuranolone and selective serotonin reuptake inhibitors for postpartum depression. J. Med. Econ. 27(1), 582–595 (2024).
59.
Swallow E, Fang A, Signorovitch J, Plumb J, Borghs S. Can matching-adjusted indirect comparison methods mitigate placebo response differences among patient populations in adjunctive trials of brivaracetam and levetiracetam? CNS Drugs 31(10), 899–910 (2017).
60.
Chen H, Qiu Y, Yin Z et al. Efficacy and safety of the innovative monoclonal antibodies in adults with generalized myasthenia gravis: a Bayesian network analysis. Front. Immunol. 14, 1280226 (2023).
61.
Ma Y, Nie X, Zhu G, Qi W, Hao L, Guo X. The efficacy and safety of different targeted drugs for the treatment of generalized myasthenia gravis: a systematic review and Bayesian network meta-analysis. CNS Drugs 38(2), 93–104 (2024).
62.
Saccà F, Pane C, Espinosa PE, Sormani MP, Signori A. Efficacy of innovative therapies in myasthenia gravis: a systematic review, meta-analysis and network meta-analysis. Eur. J. Neurol. 30(12), 3854–3867 (2023).
63.
Song Z, Zhang J, Meng J et al. Different monoclonal antibodies in myasthenia gravis: a Bayesian network meta-analysis. Front. Pharmacol. 12, 790834 (2022).
64.
Celico L, Spaepen E, De Francesco M, Iannazzo S. An indirect comparison of efgartigimod versus rituximab for generalized myasthenia gravis (abstract CO123). Value Health 25(12), S42 (2022).
65.
Van Steen C, Celico L, Spaepen E et al. Efgartigimod and ravulizumab for treating acetylcholine receptor auto-antibody-positive (AChR-Ab+) generalized myasthenia gravis: indirect treatment comparison. Adv. Ther. 41, 2486–2499 (2024).
Information & Authors
Information
Published In
Copyright
© 2025 The authors. This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 Unported License
History
Received: 4 February 2025
Accepted: 22 April 2025
Published online: 5 May 2025
Keywords:
Topics
Authors
Metrics & Citations
Metrics
Article Usage
Article usage data only available from February 2023. Historical article usage data, showing the number of article downloads, is available upon request.
Citations
How to Cite
Network connectivity, between-study heterogeneity and timepoint challenges in generalized myasthenia gravis: a feasibility assessment of indirect treatment comparisons. (2025) Journal of Comparative Effectiveness Research. DOI: 10.57264/cer-2025-0009
Export citation
Select the citation format you wish to export for this article or chapter.
Citing Literature
- Saiju Jacob, Mahmoud Hashim, Brian Hutton, Kavita Gandhi, Suzy Van Sanden, Rafal Slowik, Christopher Drudge, Antoine C. El Khoury, Mi Jun Keng, Sumeet Singh, Sindhu Ramchandren, Nils Erik Gilhus, Indirect Comparison of Nipocalimab Versus Efgartigimod and Rozanolixizumab in the Treatment of Generalized Myasthenia Gravis, Advances in Therapy, 10.1007/s12325-026-03543-1, (2026).
- José Bernardo Noronha-Matos, Carlos Sousa-Soares, Beatriz Alves-Oliveira, Liliana Carvalho, Magda Moutinho, Rafael Neiva, Paulo Correia-de-Sá, Blockage of the peri-synaptic Schwann cell nicotinic α7 receptor inhibitory drive increases the neuromuscular performance in rats with experimental autoimmune Myasthenia gravis: repurposing dipyridamole usage in myasthenics, Neuropharmacology, 10.1016/j.neuropharm.2025.110775, 284, (110775), (2026).