Optimizing clinical scientific research: the cohort intervention random sampling study with historical controls
Publication: Journal of Comparative Effectiveness Research
Abstract
Randomized controlled trials (RCTs) are regarded as the highest level of evidence in medical research, but RCTs also have their drawbacks. Over the years, several alternative study designs have been introduced to address these problems. However, many of the alternative designs are often regarded as inferior to RCTs or currently not suitable for widespread implementation due to, for example, ethical or statistical problems. Thus, there is a need for study designs that have the same level of validity as RCTs, but are also suitable for large-scale implementation. The cohort intervention random sampling study (CIRSS) with historical controls meets these requirements, by combining the strengths of abovementioned designs. The CIRSS with historical controls has the potential to optimize implementation of promising new treatments as fluidly and rapidly as possible, representing real-world clinical population. Further research is required to address the range of analyses, implementation, issues, barriers and facilitators and ethical questions related to CIRSS.
Plain language summary
Randomized controlled trials (RCTs) are regarded as best study design in medical research, but RCTs also have their drawbacks. Over the years, several alternative study designs have been introduced to address these problems. However, many of the alternative designs are often regarded as inferior to RCTs or currently not suitable for widespread implementation due to, for example, ethical or statistical problems. Thus, there is a need for study designs that have the same quality as RCTs, but are also suitable for widespread implementation. The cohort intervention random sampling study (CIRSS) with historical controls meets these requirements, by combining the strengths of abovementioned designs. The CIRSS with historical controls has the potential to optimize implementation of promising new treatments as fluidly and rapidly as possible, representing real-world clinical population. Further research is required to address the range of analyses, implementation, issues, barriers and facilitators and ethical questions related to CIRSS.
Randomized controlled trials (RCTs) are regarded as the highest level of evidence in the field of comparative effectiveness research [1,2], but RCTs also have their drawbacks [3]. Limitations include lack of external validity (generalizability) [4], high costs [5,6], selection bias due to for instance loss to follow-up and a long recruitment period [7]. Over the years, alternative study designs have been introduced or used to improve the validity of comparative effectiveness of interventions [2], such as Zelen’s designs [8], pragmatic trials [9], cohort multiple randomized controlled trials (cmRCTs) [10], and even observational studies [11]. However, many of these alternative designs are currently not considered suitable for widespread implementation due to, for example, ethical or statistical problems. Compared with RCTs, observational studies are generally quicker, less expensive, simpler to conduct and include a broader range of patients [12]. However, due to the limitations of observational studies (e.g., unmeasured confounding factors), results of these studies are often viewed with suspicion and their use is regarded as inferior to RCTs [13], while well-designed observational studies have demonstrated the ability to reproduce RCT outcomes [13–15]. Pragmatic trials have been described as alternative to RCTs to obtain real-world evidence of an intervention in clinical practice. However, they have not been without criticism, because they are mostly applied for complex treatments [16], may result in ethical issues [17,18], have less control than RCTs (e.g., comorbidities, other treatments, no unique control treatment) [19], and they frequently use cluster randomization [20,21]. In the past years, RCTs within cohorts, as alternative to or form of pragmatic trials [22,23], have been introduced to combine the strengths of both trial designs and observational studies. Most of the research using cmRCT designs has been carried out in the field of oncology [24]. In order to improve and unite scientific research and routine clinical practice, this article describes a new study design: the cohort intervention random sampling study (CIRSS) with historical controls, that also combines the strengths of both RCTs and observational cohort studies.
Current study designs: benefits & limitations
The currently most commonly used study designs for comparative effectiveness (RCT and observational cohort) as well as previously introduced alternative study designs (i.e., cmRCT and Zelen’s designs) form the foundation of our new study design. A visualization of these study designs are shown in Figure 1. Before a detailed description of the new study design is given, the benefits and limitations of these existing study designs will be described in the following five categories: recruitment and informed consent (IC), internal and external validity, ethics, statistics and logistics.
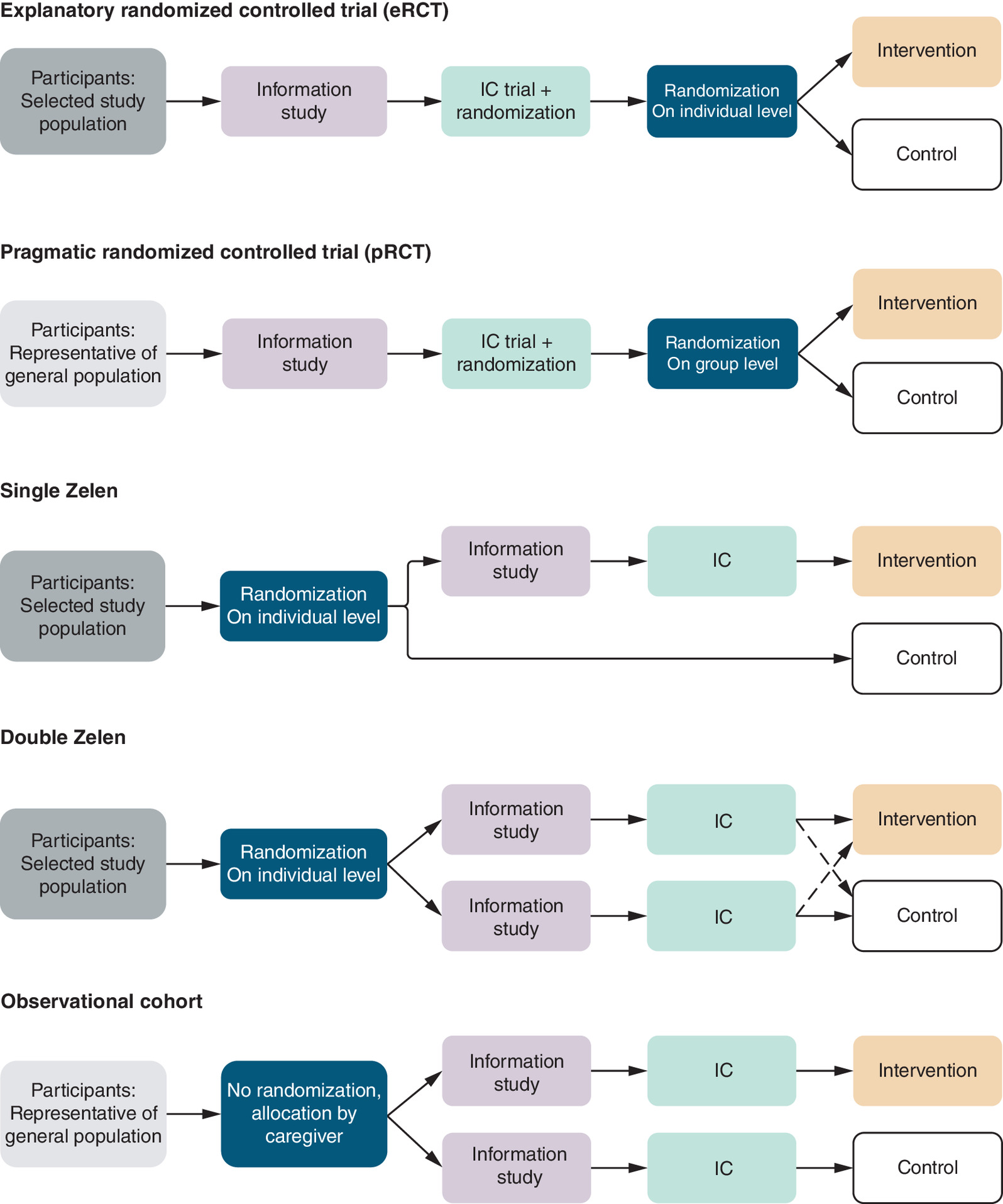
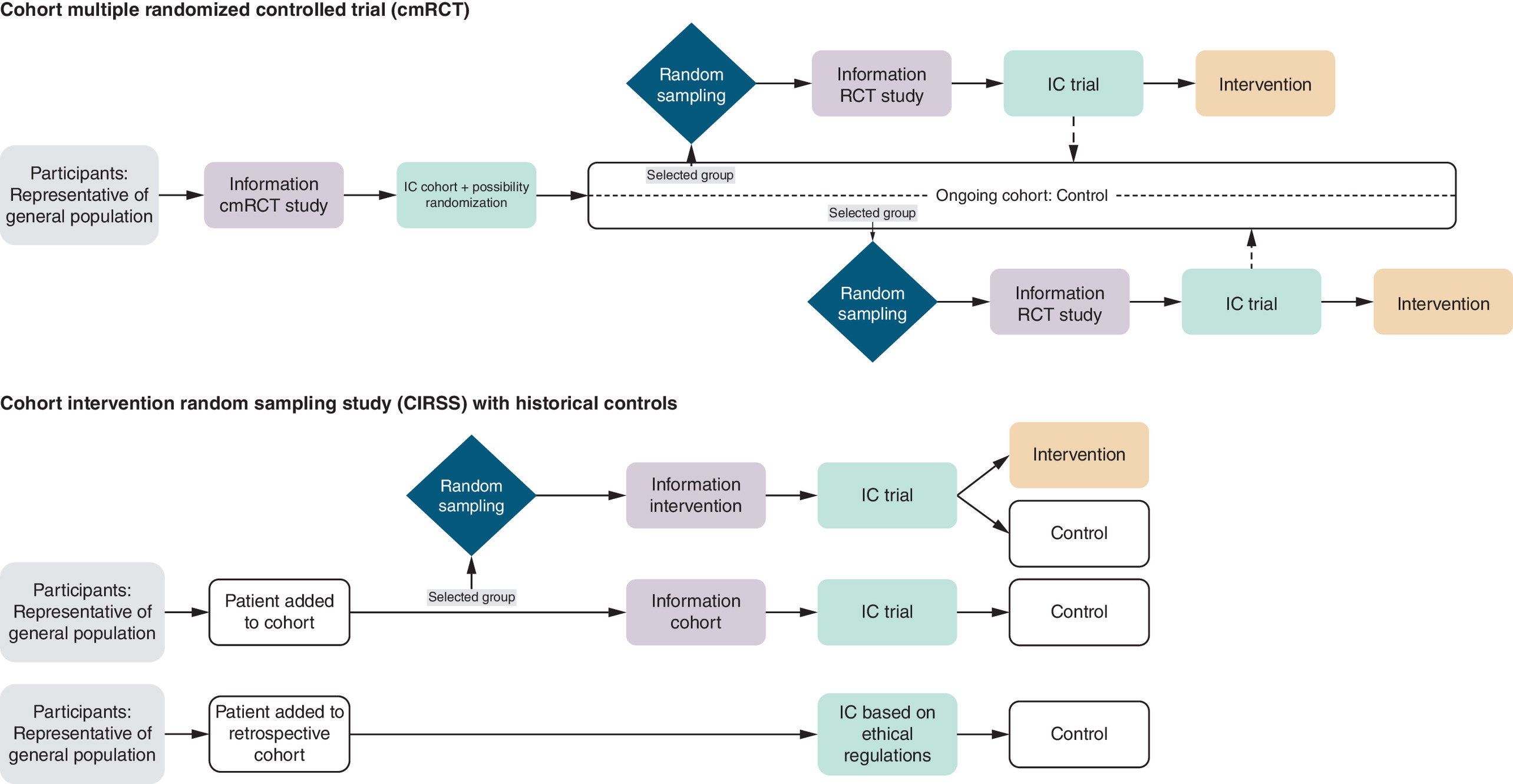
Figure 1. Recruitment and informed consent within different study designs.
IC: Informed consent; Randomized control trial.
Recruitment & IC
In explanatory and pragmatic RCTs, potential participants are informed about both the experimental and control group before randomization (pre-consent conversation). The pre-consent conversation in an RCT could potentially be an unnecessary barrier to recruitment in RCTs for both clinician and potential participant [25,26]. Clinicians may be reluctant to adequately inform the potential participant because they are hesitant to acknowledge uncertainties about alternative treatments [27] or do not (completely) inform them that the treatment they will receive is randomly chosen [10]. For participants, the decision not to participate in an RCT may stem from both a reluctance to engage in scientific research and resistance to the experimental treatment [6]. Therefore, different consent models have been suggested for pragmatic trials [26].
Furthermore, the decision to participate in the trial (apart from altruism) is to receive the experimental treatment, with a 50% certainty, because standard care is always available outside the trial [10,28]. If then placed in the control arm (the nonpreferred arm), this can result in emotional distress which can damage the physician–patient relationship [8,29], disappointment bias and noncompliance in the study arm [28,30,31].
The pre-consent conversation with abovementioned negative consequences was for the first time eliminated in Zelen’s single consent design, introduced in 1979. It is characterized by randomization without prior IC with the goal of improving clinicians’ and patients’ trial participation and to minimize bias [8]. IC for trial participation is only obtained from participants who are allocated to the experimental group. Potential participants who are allocated to the experimental treatment have 100% certainty, instead of 50% in an RCT, to receive the experimental treatment if they agree, without the uncertainty of randomization. However, the fact that control group patients are unaware of study participation is considered a major ethical concern [32,33]. Therefore, the Zelen double consent design was introduced [34]. In this ameliorated design, IC is obtained for both the experimental and control group after randomization. If a control group patient declines to receive standard care, the patient can cross over to alternative therapies, including the experimental treatment [34]. Nevertheless, Zelen’s double consent design fully eliminates the whole purpose of randomization, since each participant gets the opportunity to change to their favorite treatment.
To further ethically optimize the IC procedure, the cmRCT design was introduced [10]. This design exists of an ongoing cohort with the possibility for multiple RCTs over time. Each participant in this study design provides IC for the ongoing cohort and for potential future randomization, knowing that they might end up in an RCT. In each RCT though, only the patients randomly sampled into the intervention group and who accept this intervention, are asked to sign an additional IC.
When an RCT might be unethical or impossible, an observational cohort study is frequently performed. In a cohort study, participants, with varying exposures, are followed over a period of time and the outcome of interest is identified and treatment is allocated on the basis of what a patient needs. This follow-up can be retrospective or prospective and is sometimes combined [35]. No randomization or random sampling is applied in observational cohort studies. As a result, recruitment for observational cohort studies is often less selective and easier than for RCTs, for instance. An overview of the key differences and similarities of the study designs is given in Table 1.
| eRCT/pRCT | Single Zelen | Double Zelen | Observational Cohort | cmRCT | CIRSS with historical controls | |
|---|---|---|---|---|---|---|
| Study population | eRCT: selected study population pRCT: more representative of general population | Selected study population | Selected study population | General population when properly sampled | Representative cohort of general population with selected study population for the multiple RCT’s | General population |
| Timing recruitment and informed consent procedure | – IC before randomization – IC for study participation is obtained from all participants | – No IC before randomization – IC is only obtained from participants who are allocated to the intervention group | – No IC before randomization – IC obtained from all participants | IC from study population is obtained from all participants | – IC is obtained from all participants for the ongoing cohort and for potential future randomization – Additional IC is obtained from selected participants who are allocated to the intervention group of separate RCTs | – No IC before random selection – IC is obtained from all participants of the prospective cohort (nonsampled and sampled) – Participation in retrospective cohort with or without IC |
| Allocation method and change to receive the intervention after obtaining IC | – Randomized allocation without patient involvement in assignment – For pRCT, randomization at cluster level | Randomized allocation without patient involvement in group assignment. Participants who decline the intervention remain in their assigned group for analysis | Randomized allocation without patient involvement is changed by patient preference (crossing-over is possible) | No randomization of treatments | Randomized allocation. Participants who decline the intervention, remain part of the ongoing cohort as control | Randomized allocation in prospective cohort. Participants who decline the intervention, serve as control (after adjusting potential selection bias). N/A for the retrospective cohort |
| Probability to receive the intervention after obtaining IC | 50% (could vary a bit if randomization occurs at cluster level) | 100% | 100% | 100% | 50% | 100% for prospective cohort and 0% for retrospective cohort. |
| Statistical analysis | Intension-to-treat and/or per-protocol | Intention-to-treat | Intention-to-treat | Per-protocol | Intention-to-treat | Per-protocol |
| Ethical barriers | Participants are informed about the intervention, but have 50% chance to receive the intervention | Control group is unaware of study participation | Crossing-over can compromise the representativeness of the initially randomized population | Over collection of data compromises ethical integrity and transparency | Lack of clarity for the control group regarding the timing of the randomisation and the specific intervention | Control group is unaware of the intervention |
| Advantages of the study design | – Widely accepted and therefore easy to conduct – For eRCT confounding issues are not present – For pRCT focus on effectiveness | – Eliminate pre-consent conversation to minimize bias – Less disappointment bias – Fewer dropouts | – Eliminate pre-consent conversation to minimize bias – Less disappointment bias – Fewer dropouts, crossing-over to preferred treatment is possible | – Easy to conduct – Quicker and less expensive compared with RCTs – Multiple outcomes can be studied | – Multiple RCT’s within the same cohort – Patient-centered IC procedure – Prospective control group parallel to intervention group, makes it possible to investigate large temporal changes – Larger participation rates due to existing cohort – Focus on effectiveness | – Patient-centred IC procedure – Use of a retrospective cohort: quicker achievement of the sample size and quicker implementation of the intervention – Prospective control group parallel to intervention group, makes it possible to investigate large temporal changes – Larger participation rates due to existing cohort – Focus on effectiveness |
| Limitations of the study design | – Low external validity, but better for pRCT – Time-consuming – Expensive – Risk for selection bias and loss-to-follow-up | – Ethical concerns because control group is unaware of study participation | – Two biases due to crossing-over – Loss to follow-up if the intervention is preferable | – No control group available – Possibility of unmeasured confounding – Loss to follow-up | – Potential patients may refuse participation in the ongoing cohort because they would not like to participate in a future RCT – Designed particularly for the field of oncology – Organization efforts to manage a large ongoing cohort and multiple RCTs simultaneously | – Logistical challenges because of new study design – Need for,complex statistical analyses to address potential selection biases |
cmRCT: Cohort multiple randomized controlled trial; CIRSS: Cohort intervention random sampling study; eRCT: Explanatory randomized controlled trial; IC: Informed consent; N/A: Non applicable; pRCT: Pragmatic randomized controlled trial; RCT: Randomized controlled trial.
Internal & external validity
Although RCTs maximize their internal validity [36], many RCTs have problems with generalizability (external validity) [37]. The study population is a poor representation of the real-world clinical population since many RCTs exclude clinically important subgroups who are hard to reach by clinicians or because of narrow entry criteria [38,39]. The goal is to determine evidence that the intervention may have benefits. With the introduction of pragmatic RCTs this limitation is often eliminated, but randomization in pragmatic trials are most often executed on groups of patients, introducing potentially statistical issues (confounding factors for groups) and losing power due to the group structure (heterogeneity of groups). Due to the reduction of selection bias in the Zelen designs and the cmRCT design, the study population is more diverse and representative [10,25,34]. Likewise, well-conducted observational cohort and case–control studies also include a broad representation of the population and have shown to provide the same level of internal validity as RCTs [13–15].
Ethics
In the pre-consent conversation in RCTs, all patients are informed about the experimental treatment with 50% chance of not receiving the treatment. Although it is beneficial to provide all patients upfront information, clinical care often seems to deviate from this since patients are rarely told about treatment options that their physicians cannot guarantee to provide [40]. However, for many years it is considered ethically required to complete counseling before randomization [10]. Furthermore, randomization implies that patients are allocated and cannot choose between treatment options due to randomization, and therefore imposes ethical challenges.
Despite improvement in recruitment and external generalizability in Zelen’s single consent design, some ethical concerns have been raised [34,41]. The participants who are randomly assigned to the control group without giving IC, are unaware of being in a trial. This is resolved in Zelen’s double consent and cmRCT designs to receive IC from all the participants, although the participants in the control group for the cmRCT may not know at what moment in time the intervention is being implemented.
A cohort study design is often performed when a RCT would be impossible or unethical [42]. Compared with an RCT, observational studies have less ethical concerns since participants have 100% certainty of the treatment due to the absence of randomization. Treatment is allocated by caregiver. In some retrospective cohort studies (e.g., large cohorts where asking consent is not practicable, provided assurance that the patient’s privacy will not be compromised) IC for trial participation is not always necessary [43].
Statistics
An advantage of an RCT is that randomization reduces bias and confounding, both known and unknown [44]. Common pitfalls in statistical analyses of RCTs are managing missing data, applying the wrong statistical test, misreporting of results of the statistical analysis, and ad hoc reporting of subgroup analysis [45]. Noncompliance issues with the new treatment are typically addressed by an intention-to-treat approach. In pragmatic trials, additional (bias) issues may arise due to the cluster structure of randomization and researchers have proposed to use a per-protocol analysis to be able to estimate the effect of treatment instead of the treatment assignment [46].
The possibility of a high number of participants rejecting the intervention when they were randomly sampled for the new treatment raises a potential bias issue with the cmRCT design. In the double Zelen design, crossing over in the double consent design reduces noncompliance with the participants allocated to standard of care, but eliminates the effect of randomization, and will lead to loss to follow-up if the experimental treatment is preferable. This biases the true treatment effect compared with the single consent design. To solve these potential bias issues of the cmRCT and double Zelen design, an intention-to-treat analysis is recommended. However, research regarding trials that used the double consent design showed that 28% of the trials did not use an intention-to-treat analysis [34]. When looking at cohort designs, the analysis of data from cohort studies is often complex due to the large numbers of possible confounding variables [35], but cohort designs may offer a wider variety of collected variables and may be more representative to the population of interest.
Sample size
Calculation of an exact sample size for medical research is imperative [47,48] in particular for clinical trials. The sample size is typically calculated to achieve a minimal effect size that is considered clinically relevant. Not having the correct sample size is considered unethical [48]. Not including enough participants may risk a failure of the medical study to demonstrate the research goals. For instance, a true clinical benefit of the new treatment may not be detected due to lack-of-power of a too small sample size, while in prospective cohort studies specific associations may not be detected either. On the other hand, including too many participants may cause too much unnecessary burden on participants. This implies that too many participants may have been treated with a less beneficial treatment (depending on which treatment is clinically best), whereas a smaller trial could already have shown which treatment would be best. Also in cohorts, participant burden could be high due to the large number of variables that are being collected. Nevertheless, it should be noted that literature shows that studies are typically underpowered, because initial information is often too limited or too promising [49]. A disadvantage of clinical trials is that there are often difficulties to achieve the planned sample size due to lower than anticipated accrual rates [50]. In some cases, the clinical trial needs to be terminated before the final sample size can be achieved. In prospective cohort studies, participation rates are also declining [51,52]. However, within the cmRCT design, sample sizes may be attained more quickly due to the continuous inclusion of participants in the cohort.
Logistics
Due to RCT being the gold standard in current clinical research practices, clinicians are familiar and conversant with the logistics of the study design. As a result, the study tasks are easier adapted into clinical practice and are carried out quicker. On the other hand, the majority of RCTs initially have a large sample size to achieve the intended treatment effect. This is accompanied by a long recruitment period and high costs [5,39]. It has been shown that only 55% of the studies reach their recruitment target [53]. In addition, 45% of trials had to request for extension to reach recruitment targets [53]. Due to the long recruitment period and follow-up time of a RCT, it takes years to implement a new treatment. As a result, RCTs are less able to quickly follow clinical innovations [5]. During RCTs, treatment guidelines are sometimes modified and it is likely for the new intervention to be overtaken by a competing intervention or by a technical improved version of itself [6]. Due to time and/or monetary constraints, RCTs may also rely on surrogate markers because of feasibility, and those do not always correlate well with the outcome of interest [5].
Within the cmRCT design, numbers required for RCTs are attained more quickly because of the continuous inclusion of participants in the cohort. Also, the expenses of organizing and carrying out RCTs can be greatly decreased by centralizing the data process. Furthermore, it allows for multiple interventions to be tested simultaneously or sequentially within the same cohort [10]. The challenges of this design are the significant organizational efforts required to manage a large ongoing cohort and simultaneously coordinating multiple RCTs. Additionally, researchers, clinicians and participants in the intervention group(s) may find it difficult to comply with the multiple consent procedures.
The logistics of the Zelen design could also be doubtful. The goal of the Zelen design was to improve clinicians’ and patients’ trial participation and to minimize bias [8]. A review of trials using the Zelen design by Adamson et al. [34] described two studies with different effects on recruitment rates. Due to the poor study qualities, these results were considered questionable [34].
Data collection in a prospective cohort could also be challenging, due to the time-consuming, expensive and complex nature caused by confounders, selection-bias and loss-to-follow-up [35,54]. A retrospective cohort can be used as study design to shorten the time to complete the study and is therefore cheaper in comparison to a prospective cohort. However, additional problems such as recall bias and missing data may be associated with the use of a retrospective cohort [35,54].
The CIRSS with historical controls
We suggest a novel strategy that combines the benefits of the (cm)RCT and cohort study design with several objectives. First and foremost, the aim is to accurately reflect the real patient population and to evaluate treatment effects within this population as part of routine practice. Additionally, and equally crucial, the objective is to implement new treatments or interventions as patient-centered and effective as possible. See Figure 2 for an overview of the CIRSS, including a large prospective cohort alongside historical controls (retrospective cohort).
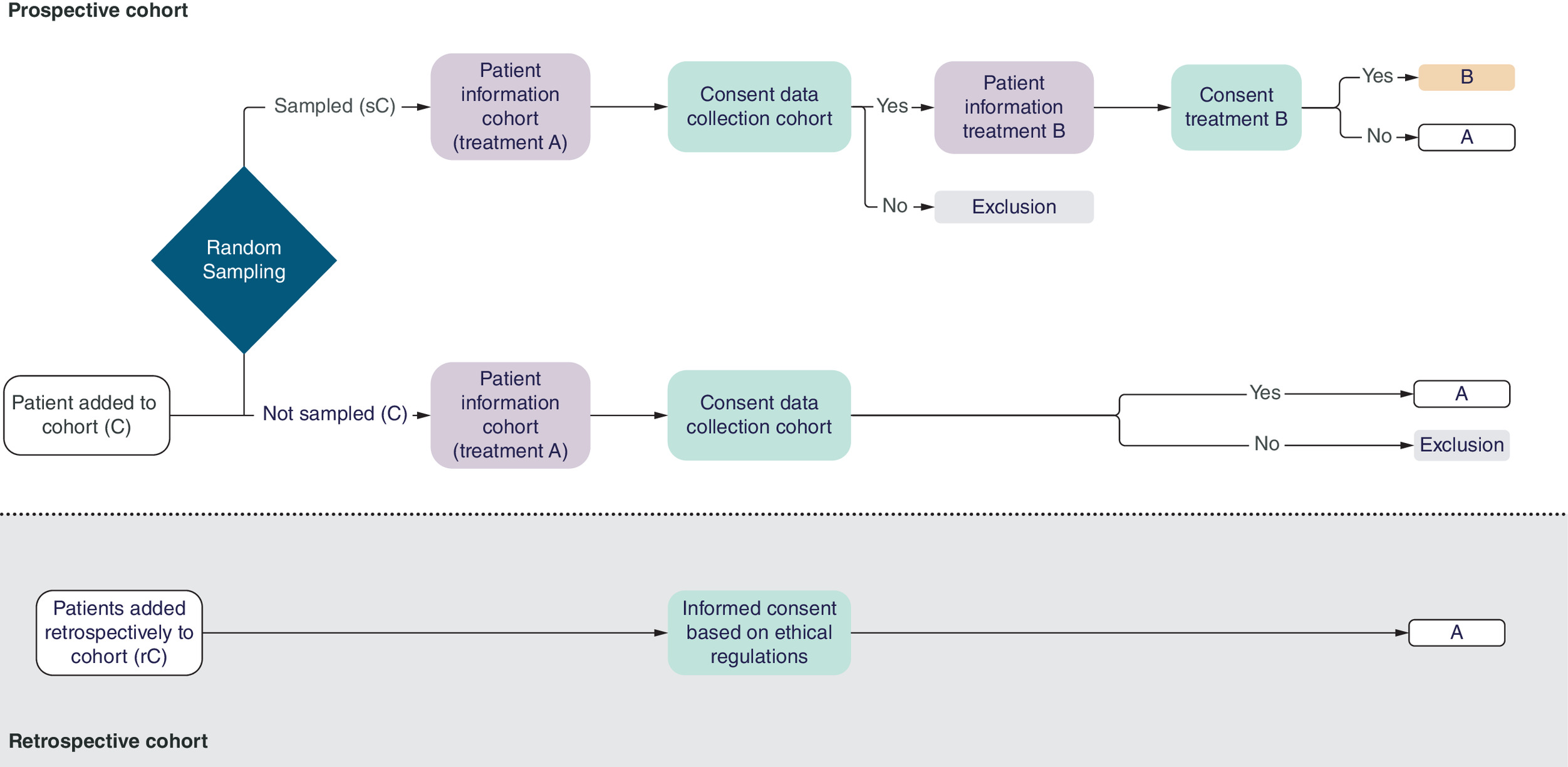
Figure 2. Cohort intervention random sampling study with historical controls.
Treatment A is generally the standard treatment, treatment B the new treatment.
Recruitment
Prospective recruitment
After the patient’s eligibility is established (i.e., any patient who would in potential be able to receive the treatment), the patient is added to the prospective cohort (C). All patients will be part of this cohort without their consent, in which treatment A (standard care) will be administered. A random sample of patients (sC) is collected from the prospective cohort (C) after which the group of randomly sampled patients will be asked to give their consent for their data to be used for research purposes (i.e., if they wish to participate in the prospective cohort). If the patient declines participation in the cohort, the patient will be excluded from the study. These patients will receive treatment A (standard care) and their data is not used. Those who agree to participate in the prospective cohort (i.e., agree to sharing data) are subsequently asked if they are willing to receive a new treatment (treatment B). All treatment options, potential benefits and risks, are explained. If the patient consents to participate in the cohort and to the new treatment (B), the new treatment (B) will be administered; if the patient consents to participate in the cohort but declines the new treatment (B), the patient will receive the standard treatment (A), and his/her data will be used in the analysis.
The group of eligible patients who are not sampled is asked to participate in the prospective cohort, and will all receive the standard treatment (A). No information concerning the new treatment (B) is provided. Those who provide consent to join the cohort, form part of the control group and will be used in the analysis. Those who decline to participate in the cohort will receive standard treatment (A) and will not submit any data.
Retrospective recruitment
In order to finish the CIRSS as soon as possible, historical controls receiving standard care (treatment A) from a retrospective cohort (rC) will be supplemented to the prospective cohort.
Results of both groups – the control group (C and rC) and the intervention group (sC) – from the prospective and retrospective cohort are then used to determine the effectiveness of the new treatment. See the Figures 1 & 2 for an overview of these groups.
The recruiting process in the CIRSS resembles the process in the cmRCT design, but there are a two exceptions. First, in CIRSS there is no consent requested for sampling, while cmRCT requests approval for sampling. This may lead to a larger control group in CIRSS than in cmRCT when participants would not like to participate in a future trial. Together with the historical controls, more patients can be immediately allocated to the new intervention (a sampling proportion beyond 50%). Having a part of the control group (the prospective control group) parallel to the intervention group, makes it possible to investigate large temporal changes. Second, the cmRCT is intended for multiple RCTs, possibly for different treatment and diseases, while CIRSS creates a cohort of patients eligible for the single intervention being studied. This implies that the control group will be better defined in CIRSS than in cmRCTs, potentially generating a less biased treatment effect.
Random sampling & patient-centered IC
The objective is to include all eligible patients in a prospective cohort that is used for both clinical evidence of standard care and intervention study at the same time. From the eligible patients, a random sampling procedure is implemented that samples patients for the new treatment. By employing this random sampling technique, prior consent is not required, unlike the standard randomization process used in RCTs. As ‘random allocation of all’ and something that is ‘done’ to all patients in RCTs, randomization is typically understood to require prior consent of the patient. The decision to receive treatment A or B in the study lies with the randomization procedure. In the CIRSS, participants in the prospective cohort (i.e., agree to sharing data) are asked if they are willing to receive treatment B when they fall in the sample. If they decline treatment B, they will receive treatment A and remain part of the study. Obviously, patients that have not been sampled, do not need IC for not being sampled. Additionally, after the random sampling procedure, IC of all sampled patients is asked. They have the full right to decline the new treatment and receive standard care within or outside of the cohort. Subsequently, the nonsampled group receives information about standard care as is done in normal practice. An overview of the key features of our study design is provided in Table 2.
| Prospective recruitment | |
| I | Recruitment of patients for a prospective observational cohort (C) |
| II | Random sampling of a number of patients (sC) from the observational cohort (C) |
| III | – Patients in the cohort (C) receive information about data collection while receiving standard treatment within the study – Patients who are randomly sampled from the cohort (sC) receive information about the standard treatment and the new treatment (intervention) in the study The trial intervention is only offered to the sampled cohort patients (sC). The patients from the cohort (C) are unaware of the intervention within the study. |
| IV | Informed consent. This process is ‘patient centered’, focusing on replicating standard care as much as possible, while trying not to overburden patients with information about nonoffered treatment. Patients in the sampled group (sC) are free to decline the intervention, and still take part in the clinical study receiving standard treatment. |
| V | Measurements of outcomes of the observational cohort (C) receiving standard treatment, and of the sampled patients from the cohort (sC) receiving the intervention (or standard treatment) during the clinical study. |
| Retrospective recruitment | |
| VI | Outcomes are gathered from a large retrospective cohort of patients receiving standard treatment (rC), to reinforce the control group from the prospective cohort. |
| Both parts combined | |
| VII | Measurements of both groups – the control group (C and rC) and the intervention group (sC) – are compared. |
Internal & external validity
We anticipate that more patients will consent to participate in the CIRSS (due to a better recruitment strategy) in comparison to – for example – RCTs [10]. There is less exclusion criteria and all cohort participants will play a role in the analysis of the study, resembling the cmRCT design. Therefore, this would most likely lead to a better representation of the general population (external validation) [10,55]. Additionally, standard care in the nonsampled group is not affected by the possibility of the use of the new treatment option and will therefore better resemble routine standard care [10]. The sampling approach aligns very much with the randomization and therefore mimics a clinical trial (internal validity), although the sampled patients are aware of the experimental setting but the control group is not.
Ethics
There are some major ethical advantages to this study design. First, by including the retrospective cohort as part of the control group, the intervention can be made available to more patients in the prospective cohort. This will lead to less burden to the patients (i.e., a smaller number prospective participants needed to reach the sample size) and a possible quicker implementation of new interventions or treatments. Second, the IC procedure is patient-centered, also contributing to as little stress as possible on the patient and caregiver. Additionally, randomly sampled patients are free to decline the intervention and have the opportunity to still take part in the study while receiving standard treatment. The sampling approach being fully random also gives every patient the same chance of being part of the intervention.
However, this CIRSS with historical controls poses a few ethical quandaries. The notion that the control group is unaware of the intervention (group) may be the leading issue, as discussed for Zelen’s single consent design, but 50 years has passed after his introduction of postrandomization and new insights, discoveries and advancements in the ethical domain may be needed. One of the compelling arguments for the uninformed control group is that this group offers a more accurate representation of reality (current clinical care). In addition, since not everyone will be sampled for the intervention, it can be more ethical to spare them from unneeded information and avoid putting them through possibly emotional distress from hearing they will not receive the intervention [28,29]. Additionally, this could avoid any potential strain on the caregiver–patient relationship, as reported in RCTs [8,29]. However, the discovery of the existence of an intervention (group) within the CIRSS by the patients in the prospective control group could lead to another ethical dilemma. The healthcare professional may find themselves in a difficult position as a result. Prior to the start of the study, this must be properly considered and its risk must be estimated.
Statistics
In the CIRSS, participants who receive the experimental treatment are typically randomly sampled from the cohort and this splits the cohort in two representative sub cohorts (of possibly different size). Like in RCTs, this also addresses the issue of confounding, in particular when all sampled participants accept to undergo the experimental treatment. Selection bias in the CIRSS may occur when the sampled participants choose the control treatment, but this seems of a similar bias problem as the selection bias that comes from noncompliance [22]. Noncompliant participants are often a nonrandom group of participants. Additionally, the selection bias due to the refusals of the experimental treatment is compensated by an unbiased estimate of the clinical effect for control treatment obtained in the nonsampled participants. These nonsampled participants (and the historical controls) use the control treatment without knowing about the existence of other experimental treatments and are therefore unaffected by their knowledge of being present in a clinical trial. This provides an opportunity to investigate the potential of selection bias by comparing the clinical outcome of the refusals with clinical outcome of the nonsampled patients, but it requires some caution since historical controls may also introduce a temporal bias due to time-related factors (e.g., different patient population, seasonal effects, other innovations that affect the clinical outcome) compared with the nonsampled participants.
The possible biases obtained in CIRSS are not unsolvable, since they can be corrected by making use of causal inference [56,57] and hybrid sampling [58] approaches specifically developed for cohort studies. Indeed, the analysis of the CIRSS with historical controls will be analyzed as a single cohort study, with one treatment group and three different control groups (a proper control, a historical control, and a group of refusals). Potential imbalances and confounders may be corrected with inverse probability weighing approaches, assuming that the possible selection and temporal biases can be corrected with the collected participant information. Thus to correct for biases, a per-protocol analysis is proposed, following the advice for pragmatic trials from causal inference researchers [46], instead of an intention-to-treat analysis. In this respect, the CIRSS also deviates from the cmRCT, where an intention-to-treat analysis is suggested [59,60], although researchers are criticizing for this approach for cmRCTs as well [58,61]. More specifically, two weighting approaches are used to correct for possible biases that may occur due to the group of patients who are offered the new treatment B but receive current treatment A (either by choice or specific circumstances), referred to as the group of refusals in CIRSS and the retrospective and prospective nature of the study, referred to as a possible temporal bias or cohort effect. Weights (based on propensity score analyses using the observed information on participants unrelated to clinical outcomes) are created to correct for imbalances in patient characteristics between the four treatment groups. First a propensity score is determined between the retrospective and prospective patients to eliminate the cohort effect from temporal differences. This will lead to an inverse probability weight for each patient in the full study. Then secondly, the calculated weights for the group of refusals are corrected by the observed patient information from the nonsampled patients in the CIRSS (i.e., the proper control group) using inverse weighting probability that is common in the field of hybrid sampling [62]. This makes the group of refusals representative in characteristics to the nonsampled control group (creating one representative control group). After establishing the weights for all patient in the full cohort study (possibly rescaled to make the weights add up to the size of the full cohort study), the clinical outcomes can be analyzed by standard weighted (logistic) regression approaches where the intervention is compared with the combined control group. Similar causal inference analyses approaches (instrumental variables) have been described for cmRCT designs, where the same type of selection may occur by the design [22,59].
Sample size
Clinical trials (both superiority and noninferiority trials) require a proper sample size calculation for a well-defined effect size, since the goal is to demonstrate a clinically relevant benefit of the new treatment. Similarly, CIRSS requires a sample size calculation when the study must empirically establish a relevant benefit for e.g., a new treatment compared with an existing treatment. The common approach is that the sample size calculation follows the proposed statistical analysis for the primary outcome. However, since the statistical analysis for CIRSS is rather complex due to the proposed data analytic approaches to accommodate potential biases. Thus, an exact sample size calculation for CIRSS, when a well-defined clinical effect is provided, is not so straightforward and requires additional research. Therefore, sample size calculation should at least be conducted under certain ideal situations, where it is assumed that selection biases are not present. The sample size calculation would then follow existing approaches for sample size calculations. Note that literature shows that covariate adjusted analyses (like the causal inference approaches proposed) in RTC’s may also lead to an increase in power [63].
Logistics
Well-conducted observational cohort and case–control studies can provide the same level of internal validity as RCTs [13–15]. Furthermore, by adding a retrospective cohort to reinforce the prospective cohort, it reduces the number of prospective patients who need to receive standard care. This relatively increases the number of patients who could benefit from the new treatment and fasten the enrolment of the required number of patients. Additionally, it could lead to a faster implementation of new treatments, preventing the new intervention to be overtaken by a competing intervention of by a technically improved version of itself. Moreover, there could be financial benefits from higher effectiveness, which are especially interesting for specific patient populations and therapies, such as expensive interventions. An applied example of this study design, including operational aspects, including infrastructure, data integration and clinical implementation, are detailed in the NIEM-II study protocol [64]. Prior to the start of a study with CIRSS design, it is recommended to inform and train care givers about the IC procedure (approaching every potential participant, separate counseling for intervention group and control group and ensure that the control group remains unaware of the intervention).
Conclusion & future perspective
The CIRSS (with historical controls) aims to unite scientific research and routine clinical practice, and optimize implementation of promising new treatments as fluid, patient-centered and rapidly as possible. CIRSS thus combine the strengths of current study designs, e.g., (pragmatic) RCTs, cmRCTs, and Zelen’s designs, aiming to overcome some of the challenges presented using these designs. The CIRSS has several distinctive features: a large observational cohort of target patients is recruited and better resembles the true population; randomly sampling participants takes place (which avoids randomization conducted in standard clinical trials); and ‘patient-centered’ information and consent is applied.
No outcome of studies using the CIRSS design are published yet. Two large clinical trials in obstetrics in the Netherlands (NIEM-II study (NCT06135961) and the NIEM-O study (NCT06151613)) using this new study design, are ongoing. Both studies received approval from the Medical Ethics Committee of Máxima MC, Veldhoven, the Netherlands with respectively number W22.071 and W22.070. The CIRSS design aligns with international research ethics guidelines (e.g., Declaration of Helsinki, principle 11, 25 and 26). As an illustration, the clinical implementation of the CIRSS design in the NIEM-II study is presented in Box 1. Further methodological details, including some statistical considerations and design rationale, are described in the corresponding study protocol [64]. CIRSS is an experimental study design in which new treatments can be evaluated against existing or current treatments. It is similar in spirit as cohort multiple randomized control trials where it shares many features of the cmRCT, but it has also some peculiarities. For instance, it allows the use of historic controls easier than cmRCT’s. In combination with proper statistical analysis from causal inference theory and hybrid sampling it is expected that an unbiased treatment effect can be obtained. The execution of CIRSS requires proper planning and organization, but in combination with historic controls, implementation of new treatments can be fast or rejected timely. We believe the CIRSS design is broadly applicable, although it might pose challenges in rare disorders, due to the necessity of large sample sizes or long accrual times. Further research is required to address the range of analyses, implementation, issues, barriers and facilitators and ethical questions related to this cohort intervention study design.
NIEM-II study
This study aims to investigate whether electrophysiological monitoring (eCTG) during labor effect the number of operative interventions, maternal and perinatal outcomes compared to conventional monitoring.
Recruitment, random sampling and informed consent
All term pregnant women in labor, admitted to Máxima Medical Center from November 2023 on, are included in the prospective cohort. A random sample with 9:1 ratio (sampled vs nonsampled) is collected. Nonsampled women receive information regarding the study, using conventional monitoring. Sampled women receive information regarding, and are offered, eCTG. They also have the opportunity to reject eCTG, while receiving conventional monitoring in or outside the study. The fetal monitoring device is applied after informed consent is obtained. Historical controls entail a large retrospective cohort of 2100 women who received conventional monitoring between 2019 and 2023. Informed consent is obtained.
Ethics
The control group being unaware of the intervention (group) was initially believed to be the leading issue. However, this wasn't apparent throughout the first part of the study.
Statistics
The study will be powered on an expected absolute reduction of operative interventions of 3% in the sampled group who will receive eCTG monitoring. To detect this difference with a power of at least 80% and a type I error of at most 5%, a total of 3471 women need to be recruited in the prospective cohort. For the retrospective cohort, a total of 2100 participants are included. Statistical methods will include mixed effects models in combination with a missing data (e.g., imputation) and propensity score approaches to correct for confounding and selection biases.
Author contributions
ND de Klerk and PBQ Berben were involved in the conceptual design, drafting, organization and overall revision of the manuscript. SG Oei, M van der Ven, AF Fransen and JOEH van Laar were involved in the conceptual design of the study and revision of the manuscript. MB van der Hout-van der Jagt was involved in the revision of the manuscript. ER van den Heuvel was involved in the conceptual design, statistical part, drafting and revision of the manuscript. All authors have read and approved the final version of the manuscript.
Financial disclosure
The authors disclosed receipt of the financial support by ZonMw – The Highly Specialized Care & Research Programme, The Hague, the Netherlands (10070022010001) and the support of Máxima Medical Center Fund. The sponsors were not involved in the conceptual design and draft of this manuscript.
Competing interests disclosure
The authors have no competing interests or relevant affiliations with any organization or entity with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Writing disclosure
No funded writing assistance was utilized in the production of this manuscript.
Open access
This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 Unported License. To view a copy of this license, visit https://creativecommons.org/licenses/by-nc-nd/4.0/
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
1.
The periodic health examination. Canadian task force on the periodic health examination. Can. Med. Assoc. J. 121(9), 1193–1254 (1979).
2.
Armstrong K. Methods in comparative effectiveness research. J. Clin. Oncol. 30(34), 4208–4214 (2012).
3.
Kostis JB, Dobrzynski JM. Limitations of randomized clinical trials. Am. J. Cardiol. 129, 109–115 (2020).
4.
Rothwell PM. External validity of randomised controlled trials: “To whom do the results of this trial apply?”. Lancet 365(9453), 82–93 (2005).
5.
Frieden TR. Evidence for health decision making — beyond randomized, controlled trials. N. Engl. J. Med. 377(5), 465–475 (2017).
•• Describes why randomized controlled trials (RCTs) are regarded as the highest level of evidence in the field of comparative effectiveness research and are therefore used for policy decisions, but also describes their drawbacks and other alternatives.
6.
Verkooijen HML, Roes K, van Gils CH. Cohort multiple randomized controlled trial: a solution for the evaluation of multiple interventions. Ned Tijdschr. Geneeskd. 157(17), A5762 (2013).
7.
McDonald AM, Knight RC, Campbell MK et al. What influences recruitment to randomised controlled trials? A review of trials funded by two UK funding agencies. Trials 7(1), 9 (2006).
8.
Zelen M. A new design for randomized clinical trials. N. Engl. J. Med. 300(22), 1242–1245 (1979).
•• Describes the Zelen’s single consent design which was the first study that eliminated the pre-consent conversation.
9.
Ford I, Norrie J. Pragmatic Trials. N. Engl. J. Med. 375(5), 454–463 (2016).
10.
Relton C, Torgerson D, O'Cathain A, Nicholl J. Rethinking pragmatic randomised controlled trials: introducing the ‘cohort multiple randomised controlled trial’ design. BMJ 340(mar19 1), c1066–c1066 (2010).
•• Describes the cohort multiple randomized controlled trial design, that further optimized the patient-centered informed consent (IC) procedure. Only the participants that were randomly sampled into the intervention group.
11.
Concato J, Lawler EV, Lew RA, Gaziano JM, Aslan M, Huang GD. Observational methods in comparative effectiveness research. Am. J. Med. 123(12), e16–e23 (2010).
12.
Feinstein AR. Epidemiologic analyses of causation: the unlearned scientific lessons of randomized trials. J. Clin. Epidemiol. 42(6), 481–489 (1989).
13.
Concato J. Observational versus experimental studies: what's the evidence for a hierarchy? NeuroRX 1(3), 341–347 (2004).
•• Describes that well-conducted observational cohort and case–control studies have shown to provide the same level of internal validity as RCTs.
14.
Concato J, Shah N, Horwitz RI. Randomized, controlled trials, observational studies, and the hierarchy of research designs. N. Engl. J. Med. 342(25), 1887–1892 (2000).
15.
Benson K, Hartz AJ. A comparison of observational studies and randomized, controlled trials. N. Engl. J. Med. 342(25), 1878–1886 (2000).
16.
Ali J, Andrews JE, Somkin CP, Rabinovich CE. Harms, benefits, and the nature of interventions in pragmatic clinical trials. Clinical Trials 12(5), 467–475 (2015).
17.
Nicholls SG, Carroll K, Zwarenstein M et al. The ethical challenges raised in the design and conduct of pragmatic trials: an interview study with key stakeholders. Trials 20(1), 765 (2019).
18.
Goldstein CE, Weijer C, Brehaut JC et al. Ethical issues in pragmatic randomized controlled trials: a review of the recent literature identifies gaps in ethical argumentation. BMC Med. Ethics 19(1), 14 (2018).
19.
Roland M, Torgerson DJ. Understanding controlled trials: what are pragmatic trials? Br. Med. J. 316(7127), 285–285 (1998).
20.
Hemming K, Taljaard M. Key considerations for designing, conducting and analysing a cluster randomized trial. Int. J. Epidemiol. 52(5), 1648–1658 (2023).
21.
Taljaard M, Goldstein CE, Giraudeau B et al. Cluster over individual randomization: are study design choices appropriately justified? Review of a random sample of trials. Clinical Trials 17(3), 253–263 (2020).
22.
van der Velden JM, Verkooijen HM, Young-Afat DA et al. The cohort multiple randomized controlled trial design: a valid and efficient alternative to pragmatic trials? Int. J. Epidemiol. 46(1), 96–102 (2017).
23.
Kim SY, Flory J, Relton C. Ethics and practice of trials within cohorts: an emerging pragmatic trial design. Clinical Trials 15(1), 9–16 (2018).
24.
Nickolls BJ, Relton C, Hemkens L et al. Randomised trials conducted using cohorts: a scoping review. BMJ Open 14(3), e075601 (2024).
25.
Homer CSE. Using the Zelen design in randomized controlled trials: debates and controversies. J. Adv. Nurs. 38(2), 200–207 (2002).
26.
Kalkman S, van Thiel GJMW, Zuidgeest MGP et al. Series: pragmatic trials and real world evidence: paper 4. Informed consent. J. Clin. Epidemiol. 89, 181–187 (2017).
27.
Simon GE, Shortreed SM, DeBar LL. Zelen design clinical trials: why, when, and how. Trials 22(1), 541 (2021).
28.
Bradley C. Designing medical and educational intervention studies: a review of some alternatives to conventional randomized controlled trials. Diabetes Care 16(2), 509–518 (1993).
29.
Flory JH, Mushlin AI, Goodman ZI. Proposals to conduct randomized controlled trials without informed consent: a narrative review. J. Gen. Intern. Med. 31(12), 1511–1518 (2016).
30.
Schellings R, Kessels AGH, ter Riet G, Sturmans F. The Zelen design may be the best choice for a heroin-provision experiment. J. Clin. Epidemiol. 52(6), 503–507 (1999).
31.
Victora CG, Habicht J-P, Bryce J. Evidence-based public health: moving beyond randomized trials. Am. J. Public Health 94(3), 400–405 (2004).
32.
Ellenberg SS. Informed consent: protection or obstacle? Some emerging issues. Control. Clin. Trials 18(6), 628–636 (1997).
33.
Beauchamp TL, Cook RR, Fayerweather WE et al. Ethical guidelines for epidemiologists. J. Clin. Epidemiol. 44, 151–169 (1991).
34.
Adamson J, Cockayne S, Puffer S, Torgerson DJ. Review of randomised trials using the post-randomised consent (Zelen's) design. Contemp. Clin. Trials 27(4), 305–319 (2006).
35.
Barrett D, Noble H. What are cohort studies? Evidence Based Nursing 22(4), 95–96 (2019).
36.
Kennedy-Martin T, Curtis S, Faries D, Robinson S, Johnston J. A literature review on the representativeness of randomized controlled trial samples and implications for the external validity of trial results. Trials 16(1), 495 (2015).
37.
Tan YY, Papez V, Chang WH, Mueller SH, Denaxas S, Lai AG. Comparing clinical trial population representativeness to real-world populations: an external validity analysis encompassing 43 895 trials and 5 685 738 individuals across 989 unique drugs and 286 conditions in England. Lancet Healthy Longev. 3(10), e674–e689 (2022).
38.
Mason S. Representation of South Asian people in randomised clinical trials: analysis of trials' data. BMJ 326(7401), 1244–1245 (2003).
39.
Luce BR. Rethinking randomized clinical trials for comparative effectiveness research: the need for transformational change. Ann. Intern. Med. 151(3), 206 (2009).
•• Describes that many RCTs use strict inclusion and exclusion criteria. The study results are not representative to reflect the circumstances used in common clinical practice.
40.
Firth J. Should you tell patients about beneficial treatments that they cannot have? No. BMJ 334(7598), 827–827 (2007).
41.
Zelen M. Randomized consent designs for clinical trials: an update. Stat. Med. 9(6), 645–656 (1990).
42.
Gilmartin-Thomas JF, Liew D, Hopper I. Observational studies and their utility for practice. Aust. Prescr. 41(3), 82–85 (2018).
43.
Health. WM of. National Ethics Advisory Committee. (2012). Ethical guidelines for observational studies: observational research, audits and related activities. (2012). http://www.neac.health.govt.nz
• Describes that IC for trial participation is not always necessary.
44.
Moher D, Hopewell S, Schulz KF et al. CONSORT 2010 explanation and elaboration: updated guidelines for reporting parallel group randomised trials. Int. J. Surg. 10(1), 28–55 (2012).
45.
Charan J, Saxena D. Suggested statistical reporting guidelines for clinical trials data. Indian J. Psychol. Med. 34(1), 25–29 (2012).
46.
Hernán MA, Robins JM. Per-protocol analyses of pragmatic trials. N. Engl. J. Med. 377(14), 1391–1398 (2017).
47.
Sathian B, Sreedharan J, Baboo SN, Sharan K, Abhilash ES, Rajesh E. Relevance of sample size determination in medical research. Nepal J. Epidemiol. 1(1), 4–10 (1970).
48.
Bacchetti P, Wolf LE, Segal MR, McCulloch CE. Ethics and sample size. Am. J. Epidemiol. 161(2), 105–10 (2005). Erratum in: Am. J. Epidemiol. 162(12), 1237 (2005).
49.
Vickers AJ. Underpowering in randomized trials reporting a sample size calculation. J. Clin. Epidemiol. 56(8), 717–720 (2003).
50.
Haidich A-B, Ioannidis JPA. Patterns of patient enrollment in randomized controlled trials. J. Clin. Epidemiol. 54(9), 877–883 (2001).
51.
Galea S, Tracy M. Participation rates in epidemiologic studies. Ann. Epidemiol. 17(9), 643–653 (2007).
52.
Keeble C, Baxter PD, Barber S, Law GR. Participation rates in epidemiology studies and surveys: a review 2007–2015. Internet J. Epidemiol. 14(1), 1–14 (2015).
53.
Sully BGO, Julious SA, Nicholl J. A reinvestigation of recruitment to randomised, controlled, multicenter trials: a review of trials funded by two UK funding agencies. Trials 14, 166 (2013).
54.
Mann CJ. Observational research methods. Research design II: cohort, cross sectional, and case-control studies. Emerg. Med. J. 20(1), 54–60 (2003).
55.
Knottnerus JA, Dinant GJ. Medicine based evidence, a prerequisite for evidence based medicine. BMJ 315(7116), 1109–1110 (1997).
56.
Rubin DB. Causal inference using potential outcomes: design, modeling, decisions. J. Am. Stat. Assoc. 100, 322–331 (2005).
57.
Little RJ, Rubin DB. Causal effects in clinical and epidemiological studies via potential outcomes: concepts and analytical approaches. Annu. Rev. Public Health 21(1), 121–145 (2000).
58.
Pate A, Candlish J, Sperrin M, Van Staa TP. Cohort multiple randomised controlled trials (cmRCT) design: efficient but biased? A simulation study to evaluate the feasibility of the cluster cmRCT design. BMC Med. Res. Methodol. 16(1), 109 (2016).
59.
Narzari H, Nilima N, Vishnu VY, Khan MA, Gupta A, Srivastava VP. A systematic review of the statistical methods adopted for analyzing follow-up data in cohort multiple randomized controlled trial. Cureus 16(1), e51558 (2024).
60.
Viksveen P, Relton C, Nicholl J. Benefits and challenges of using the cohort multiple randomised controlled trial design for testing an intervention for depression. Trials 18(1), 308 (2017).
61.
Candlish J, Pate A, Sperrin M, van Staa T. Evaluation of biases present in the cohort multiple randomised controlled trial design: a simulation study. BMC Med. Res. Methodol. 17(1), 17 (2017).
62.
Wu C. Statistical inference with non-probability survey samples. Surv. Methodol. 48(2), 313–318 (2022).
63.
Hernández AV, Steyerberg EW, Habbema JDF. Covariate adjustment in randomized controlled trials with dichotomous outcomes increases statistical power and reduces sample size requirements. J. Clin. Epidemiol. 57(5), 454–460 (2004).
64.
Berben PBQ, de Klerk ND, van der Ven M et al. Non-invasive electrophysiological monitoring vs conventional monitoring during labour in a tertiary obstetric care centre in the Netherlands: study protocol of a cohort intervention random sampling study (NIEM-II study). BMJ Open 15(7), e102901 (2025).
Information & Authors
Information
Published In
Copyright
© 2025 The authors. This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 Unported License
History
Received: 29 August 2024
Accepted: 28 August 2025
Published online: 16 September 2025
Keywords:
Topics
Authors
Metrics & Citations
Metrics
Article Usage
Article usage data only available from February 2023. Historical article usage data, showing the number of article downloads, is available upon request.
Citations
How to Cite
Optimizing clinical scientific research: the cohort intervention random sampling study with historical controls. (2025) Journal of Comparative Effectiveness Research. DOI: 10.57264/cer-2024-0168
Export citation
Select the citation format you wish to export for this article or chapter.
Citing Literature
- Phebe B. Q. Berben, Yannick L. Smolders, Ivar R. de Vries, Nadine D. de Klerk, Annemarie F. Fransen, Marta Regis, M. Beatrijs van der Hout – van der Jagt, Myrthe van der Ven, Alexandra M. J. V. Schyns‐van den Berg, S. Guid Oei, Judith O. E. H. van Laar, The effect of labor epidural analgesia on uterine activity using electrohysterography monitoring: A follow‐up study, Acta Obstetricia et Gynecologica Scandinavica, 10.1111/aogs.70187, (2026).
- Nadine D de Klerk, Phebe B Q Berben, Ivar R de Vries, Hendrik Niemarkt, Rik Vullings, Edwin R van den Heuvel, Myrthe van der Ven, Annemarie F Fransen, S Guid Oei, Judith O E H van Laar, Continuous non-invasive electrophysiological monitoring in high-risk pregnancies: study protocol of a cohort intervention random sampling study in a tertiary obstetrical care centre in the Netherlands (NIEM-O study), BMJ Open, 10.1136/bmjopen-2025-102732, 15, 11, (e102732), (2025).