Comparative effectiveness analysis between entrectinib clinical trial and crizotinib real-world data in ROS1+ NSCLC
Publication: Journal of Comparative Effectiveness Research
Abstract
Aim: Generating direct comparative evidence in prospective randomized trials is difficult for rare diseases. Real-world cohorts may supplement control populations. Methods: Entrectinib-treated adults with advanced ROS1 fusion-positive NSCLC (n = 94) from Phase I/II trials (ALKA-372-001 [EudraCT2012-00148-88], STARTRK-1 [NCT02097810], and STARTRK-2 [NCT02568267]) were compared with a real-world crizotinib-treated cohort (n = 65). Primary end point, time-to-treatment discontinuation (TTD); secondary end points, PFS and OS. Results: Median (95% CI) weighted TTD: 12.9 (9.9–17.4) months for entrectinib; 8.2 (6.2–9.9) months for crizotinib (weighted hazard ratio: 0.72 [0.51–1.02]). Median OS with entrectinib was not reached, weighted median OS with crizotinib was 18.5 (15.1–47.2) months. Conclusion: Entrectinib administered in clinical trials may be associated with longer TTD than a real-world crizotinib population.
Non-small-cell lung cancer (NSCLC) is a heterogeneous disease characterized by a range of genetic mutations driving tumorigenesis, including those in ALK, EGFR and ROS1 genes [1,2]. The ROS1 gene encodes a protein that is a receptor tyrosine kinase and is involved in recurrent chromosomal rearrangements that define a class of oncogenic driver alterations in NSCLC [3,4]. ROS1 gene fusions occur in 1–2% of NSCLC patients [5,6,7].
Entrectinib (ROZLYTREK™) is an oral tyrosine kinase inhibitor of ROS1, TRK, and ALK, that has demonstrated activity across various tumor types and histologies, including ROS1 fusion-positive (ROS1+) NSCLC [8,9,10]. Entrectinib received US FDA approval in August 2019 for the treatment of patients with ROS1+ NSCLC. In preclinical studies, entrectinib had the ability to cross the blood–brain barrier and remain within the CNS [10]. The efficacy of entrectinib was assessed in two Phase I and one Phase II single-arm studies [8,11].
Given the rarity of ROS1 fusions, a prospective randomized trial in this population is not feasible within a reasonable time-frame. To generate comparative efficacy evidence for entrectinib versus crizotinib (the current standard of care in this setting), we built a comparable cohort of ROS1+ NSCLC patients treated with crizotinib from the Flatiron Health (FH) electronic health record (EHR)-derived database to compare the clinical outcomes of entrectinib-treated trial patients with patients treated with crizotinib in routine clinical practice.
Patients & methods
Study design & population
This was a non-interventional study with secondary use of observational and clinical trial data. We compared two cohorts of patients with ROS1+ NSCLC. The entrectinib cohort included adults who received entrectinib from three open-label, single-cohort, Phase I/II trials: ALKA-372-001 (EudraCT2012-00148-88; n = 9), STARTRK-1 (NCT02097810; n = 7) and STARTRK-2 (NCT02568267; n = 78) [8,12,13]. All studies reported in this secondary analysis were funded by F Hoffmann-La Roche Ltd and were conducted in accordance with the principles of the Declaration of Helsinki and Good Clinical Practice guidelines and all patients provided written informed consent. Protocols were approved by the relevant institutional review boards and/or ethics committees.
Patient data for the crizotinib cohort were generated using the FH EHR-derived database, a USA nationwide longitudinal, demographically and geographically diverse de-identified database derived from EHR data, curated via technology-enabled abstraction [14,15]. At the time of the study, FH included de-identified data from over 265 cancer clinics (∼800 sites of care) representing more than 2 million USA cancer patients available for analysis.
Patients aged ≥18 years with advanced or metastatic (stage IIIB/IV) NSCLC at trial enrollment were included (clinical cut-off, 1 May 2019; enrollment cut-off, 30 November 2017). From the FH data, patients diagnosed with advanced NSCLC and ROS1+ rearrangements from 1 January 2011 to 30 June 2018 were included (clinical cut-off, 31 March 2020). The latest available clinical cut-off was used for each of the two datasets. All patients had a documented ROS1 fusion confirmed by next-generation sequencing (NGS) or other nucleic acid-based diagnostic tests in the entrectinib cohort, and by NGS or fluorescence in situ hybridization in the crizotinib cohort.
Patients in the crizotinib cohort were excluded if the duration between diagnosis of advanced disease or biomarker specimen date and the start of crizotinib treatment was >90 days, or if no information was available on prior crizotinib treatment. To match exclusion criteria in the entrectinib trials, patients were excluded from the crizotinib cohort if they tested positive for concomitant driver mutations (e.g., EGFR, ALK, KRAS, BRAF). The trial only included patients with an Eastern Cooperative Oncology Group performance status (ECOG-PS) of 0–2. ECOG-PS is frequently missing when using real-world data (RWD); patients with missing ECOG-PS in the crizotinib cohort were included in the analysis, but where data were available, patients were excluded if their ECOG-PS was >2.
Prior anticancer therapy before starting entrectinib or crizotinib was allowed. Patients in the study were excluded if they had prior exposure to another ROS1 inhibitor (including crizotinib), either in a real-world setting or in a clinical study. Two patients with prior crizotinib exposure were included in the integrated efficacy analysis of the original entrectinib trial [13]; these patients were also allowed in this analysis for consistency. In the crizotinib cohort, only patients who received crizotinib for the first time as monotherapy were allowed (any line of treatment).
Objectives
The primary objective was a comparative effectiveness analysis of time-to-treatment discontinuation (TTD) between entrectinib and crizotinib. Secondary objectives included a comparison between entrectinib and crizotinib with respect to overall survival (OS) and progression-free survival (PFS; captured differently for real-world and clinical trial populations; exploratory objective).
Outcome assessments
As treatment beyond progression (TBP) in clinical practice is often accompanied by local ablative therapy [16,17], the earliest progression event (including death) or treatment discontinuation in both cohorts was used to estimate TTD. In the entrectinib cohort, TTD was the date from entrectinib initiation to treatment discontinuation due to death, toxicity, consent withdrawal or progression. TTD was selected as the primary end point because it is an outcome that integrates both the patient's and the clinician's judgement of the tolerability, safety and effectiveness of a treatment. This end point was previously identified as a pragmatic end point for use in retrospective real-world evidence studies, where scans are performed at variable intervals and post-hoc objective radiological review is not always feasible. It is also an appropriate end point to use for targeted therapies, as discontinuation due to toxicity is less common in trials of targeted therapies compared with chemotherapies [18].
In the clinical trial, patients were scanned every 8 weeks and blinded independent central review (BICR)-Response Evaluation Criteria in Solid Tumors (RECIST; v1.1) was used to confirm progression. Additional analyses used investigator-assessed progression, which was also available in the entrectinib trial data. PFS in the entrectinib cohort was the time from first entrectinib dose to first documentation of radiographic disease progression (per BICR-RECIST or investigator assessment), or death due to any cause, whichever occurred first.
In the crizotinib cohort, treatment was administered according to clinical practice. Treatment start and stop dates were collected using technology-enabled abstraction for EHR-derived information. TTD was defined as the time from crizotinib initiation to treatment discontinuation. Treatment discontinuation was assumed to have occurred if there was a subsequent line of therapy, if the patient died ≤7 days from the last treatment administration, if the patient had no subsequent therapy line but the duration between the last date of crizotinib treatment and the last visit date was ≥60 days or because of disease progression. In FH, the progression variable included in TTD and PFS analyses was derived differently than in the clinical trials and is termed real-world progression (rwP). The date of rwP was based on abstraction of the treating clinician's assessment using the EHR, defined in this study as a distinct episode in which the clinician concluded evidence of tumor growth (i.e., by scans, pathology and/or clinician determination), loss of clinical benefit or both [19,20].
OS was defined as time from the date of treatment initiation until death from any cause in both cohorts, using a previously described real-world mortality variable [21]. Censoring rules are summarized in Supplementary Table 1 and page 1 in the Supplementary Material.
Statistical methods
Descriptive analyses included comparison of baseline characteristics between the entrectinib and crizotinib cohorts to identify possible differences in demographic and clinical characteristics. Baseline was defined as characteristics at enrollment for the entrectinib cohort or the closest visit/information before treatment start for the crizotinib cohort, at a maximum of 90 days prior to treatment start (index date). For BMI and ECOG, given the potential rapid changes in this type of population, the values reported were obtained within 30 days of the start of crizotinib treatment. Baseline characteristics were compared between the cohorts using Chi-square tests for categorical variables and Wilcoxon rank-sum tests for continuous variables.
The date from initiation of treatment to the first evidence of rwP was used as the discontinuation date when TBP was observed in the crizotinib cohort. Similarly, the entrectinib clinical trial patients were counted as discontinuation at the time of first progression, even in cases where TBP was allowed.
A comparison of TTD and PFS was performed after adjusting for differences between the entrectinib and crizotinib cohorts using the Inverse Probability of Treatment Weighting (IPTW) method [22]. Propensity score (PrS) was estimated by unconditional logistic regression (Supplementary Table 2). Additional details on statistical methods are provided in the Supplementary Material pages 1–2.
The clinical profile of patients with ROS1-rearranged NSCLC is similar to that of ALK-rearranged NSCLC, including early age of onset and non-smoking history [5]. Due to limitations with identifying prognostic factors from available ROS1 patient data, it was infeasible to collect data for all potential prognostic elements and these factors were selected based on the known prognostic factors for ALK-rearranged NSCLC and expert clinical judgment.
Prior therapy was derived from trial records for the entrectinib cohort and abstracted manually from clinical notes for the crizotinib cohort. Only treatments given for metastatic disease or after metastatic diagnoses were considered. Several sensitivity analyses were conducted in order to test the robustness of results (Supplementary Material page 1). Median TTD and PFS were also estimated by CNS status at baseline in both cohorts as secondary analyses.
Results
Patients
The primary analysis included 94 patients in the entrectinib cohort from the three entrectinib clinical trials [13] (Figure 1A) and 65 in the crizotinib cohort derived from a group of 134 patients with ROS1+ NSCLC identified in the FH database (Figure 1B). Patients from the entrectinib clinical trials were located in Europe, Australia, Asia and the USA. The FH database included only patients treated in the USA.
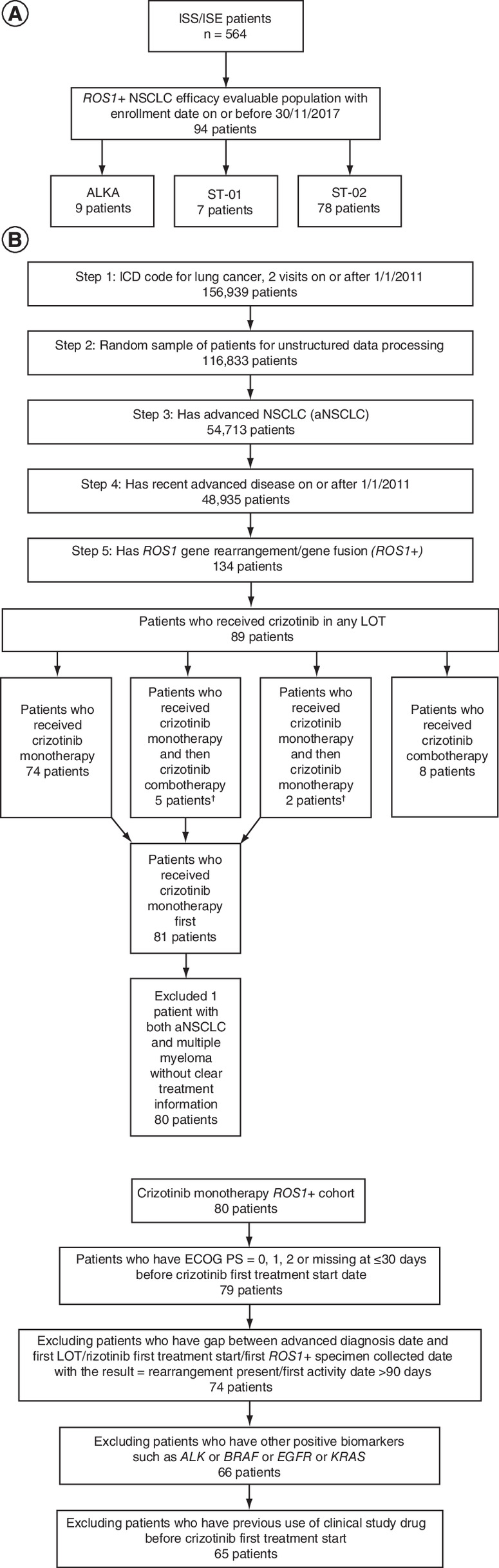
Figure 1. Patient flow diagram in the two study cohorts.
(A) Entrectinib cohort. (B) Crizotinib cohort.
†First-line treatment with crizotinib monotherapy, second-line treatment with combination therapy (crizotinib + chemotherapy) or crizotinib monotherapy.
ICD: International Statistical Classification of Diseases and Related Health Problems; ISE: Integrated summary of effectiveness; ISS: Integrated summary of safety; LOT: Line of therapy; NSCLC: Non-small-cell lung cancer.
Median age was 53 and 65 years in the entrectinib and crizotinib cohorts, respectively, and the majority of patients in both cohorts were women (Table 1). Compared with the crizotinib cohort, the entrectinib cohort included more patients of Asian ethnicity and fewer patients with a smoking history. Patients in the entrectinib cohort had a higher baseline prevalence of CNS metastases, had a higher tumor burden (≥2 baseline metastatic sites), and were more heavily pretreated (>2 prior lines of therapy) compared with patients in the crizotinib cohort. In the crizotinib cohort, 57% (37/65) of patients received TBP (Supplementary Figure 1).
| Characteristics | Entrectinib cohort (N = 94) | Crizotinib cohort (N = 65) |
|---|---|---|
| Men, n (%) | 34 (36) | 28 (43) |
| Race, n (%) | ||
| –White | 46 (49) | 35 (54) |
| – Asian | 41 (44) | 6 (9) |
| – Black/African American | 4 (4) | 8 (12) |
| – Hispanic/Latino | 1 (1) | 11 (17) |
| – Not provided | 2 (2) | 3 (5) |
| Age (years) | ||
| – 18–34, n (%) | 7 (7) | 0 |
| – 35–64, n (%) | 67 (71) | 31 (48) |
| – ≥65, n (%) | 20 (21) | 34 (52) |
| – Median (IQR) | 53 (45–62) | 65 (55–73) |
| Smoking status, n (%) | ||
| – Smoker | 38 (40) | 37 (57) |
| – Never smoker | 56 (60) | 28 (43) |
| Clinical practice type, n (%) | ||
| – Community | – | 50 (77) |
| – Academic | 94 (100) | 15 (23) |
| Location, n (%) | ||
| – Asia Pacific | 43 (46) | 0 |
| – Europe | 26 (28) | 0 |
| – USA | 25 (27) | 65 (100) |
| Histology, n (%) | ||
| – Non-squamous cell carcinoma | 94 (100) | 61 (94) |
| – Squamous cell carcinoma | 0 | 2 (3) |
| – NOS | 0 | 2 (3) |
| ECOG-PS†, n (%) | ||
| – 0 | 35 (37) | 19 (29) |
| – 1 | 48 (51) | 11 (17) |
| – 2 | 11 (12) | 7 (11) |
| – Missing | 0 | 28 (43) |
| Brain metastases at baseline, n (%) | 40 (43) | 17 (26) |
| With ≤2 metastatic sites (any site), n (%) | 50 (53) | 54 (83) |
| Prior therapy before entrectinib/crizotinib start, n (%) | ||
| – <2 | 70 (74) | 60 (92) |
| – ≥2 | 24 (26) | 5 (8) |
| Prior targeted therapy before entrectinib/crizotinib start (any line), n (%) | 13 (14) | 9 (14) |
| Prior chemotherapy before entrectinib/crizotinib start (any line), n (%) | 67 (71) | 20 (31) |
†
Assessed within the 30 days before the first treatment start date for the crizotinib cohort.
ECOG-PS: Eastern Cooperative Oncology Group performance status; IQR: Interquartile range; NOS: Not otherwise specified.
Comparative effectiveness
Seventy-one (76%) in the entrectinib cohort and 65 (84%) in the crizotinib cohort (weighted) discontinued treatment. Median TTD was 12.9 months (95% CI: 9.9–17.4 in the entrectinib cohort and 8.2 months (6.2–9.9) in the crizotinib cohort (weighted; hazard ratio [HR]: 0.72; 95% CI: 0.51–1.02) (Figure 2A; Table 2; Figure 3). Patients in the entrectinib cohort had a minimum of 12 months of follow-up after first dose (median follow-up: 19.3 months); patients in the crizotinib cohort had a median follow-up of 14.1 months (varying follow-up times were included in the crizotinib cohort to limit further reduction of sample size).

Figure 2. Kaplan–Meier estimates of the primary and secondary endpoints in the crizotinib and entrectinib cohorts.
(A) TTD, (B) PFS and (C) OS. All curves are weighted based on the propensity score for comparability between arms.
HR: Hazard ratio; OS: Overall survival; PFS: Progression-free survival; TTD: Time-to-treatment discontinuation.
| Entrectinib | Crizotinib unweighted | Crizotinib weighted | |
|---|---|---|---|
| Median TTD†, months | 12.9 | 7.7 | 8.2 |
| – (95% CI) | (9.9–17.4) | (4.9–10.0) | (6.2–9.9) |
| – n/N | 71/94 | 54/65 | 65/77 |
| Median PFS†, months | 16.8 | 7.7 | 8.2 |
| – (95% CI) | (12.0–26.3) | (5.4–10.0) | (6.5–9.9) |
| – n/N | 54/94 | 54/65 | 65/77 |
| Median OS, months | NE | 19.9 | 18.5 |
| – (95% CI) | – | (14.4–NE) | (15.1–47.2) |
| – n/N | 25/94 | 35/65 | 48/77 |
†
Progression determined by BICR in the entrectinib cohort, and from clinician report in the crizotinib cohort.
BICR: Blinded independent central review; NE: Not estimable; OS: Overall survival; PFS: Progression-free survival; TTD: Time-to-treatment discontinuation.

Figure 3. Multivariate Cox model of treatment effect.
(A) TTD. (B) PFS.
*Adjusted according to age, race, gender, smoking status, presence of brain metastases and exposure to prior lines of therapy.
PFS: Progression-free survival; TTD: Time-to-treatment discontinuation.
Fifty-four patients (57%) in the entrectinib cohort and 65 (84%) in the crizotinib cohort (weighted) had a progression or death event. Estimated median PFS determined from clinical trial data was 16.8 months (95% CI: 12.0–26.3) in the entrectinib cohort (Figure 2B; Table 2; Figure 3). Estimated median real-world PFS was 8.2 months (95% CI: 6.5–9.9) in the crizotinib cohort (weighted; HR: 0.54; 95% CI: 0.38–0.79).
Median OS could not be estimated in the entrectinib cohort because an insufficient number of events had occurred at time of data cut-off. In the crizotinib cohort, estimated median OS was 18.5 months (95% CI: 15.1–47.2) (Figure 2C; Table 2).
When using progression by investigator-assessed definition rather than BICR in the entrectinib trials, the weighted HR was 0.78 (95% CI: 0.55–1.09) and 0.70 (95% CI: 0.49–0.99) for TTD and PFS, respectively (Supplementary Table 3).
The adjusted HR for TTD and PFS did not change when patients with events within 1 or 3 months of treatment start were excluded from the analyses (Supplementary Table 4).
Sensitivity analysis of TTD
HR maintained robustness across all sensitivity analyses (Supplementary Table 5). When including only patients with non-missing ECOG-PS, HR for TTD was 0.68 (95% CI: 0.48–0.98; weighted analysis). Results using caliper matching by the PrS presented a slightly less favorable HR (0.86; 95% CI: 0.46–1.61) than the main analysis, and given the reduced sample size (N = 80, both cohorts), the 95% CI was wider. In the main analysis and the sensitivity analysis, the standardized mean difference (SMD) on weighted cohorts was close to or below 0.2 for all variables used to derive a PrS (based on the main analysis model).
Secondary analyses of CNS metastases at baseline
At treatment start, 17 (26.2%) patients in the crizotinib cohort and 40 (42.6%) patients in the entrectinib cohort had brain metastases. By CNS status, unadjusted TTD and PFS were lower in the crizotinib cohort than in the entrectinib cohort (Supplementary Table 6). In the crizotinib cohort, unadjusted median TTD was 7.9 months (95% CI: 5.6–10.8) in patients with no brain metastasis at treatment start and 5.4 months (95% CI: 2.0–13.3) in patients with brain metastasis at treatment start. In the entrectinib cohort, unadjusted median TTD was 17.1 months (95% CI: 10.5–29.6) in patients with no brain metastasis at treatment start and 7.8 months (95% CI: 4.6–15.0) in patients with brain metastasis at treatment start. After adjusting for the same a priori prognostic factors used to develop the propensity score, except for brain metastasis, the HR showed a similar benefit with entrectinib versus crizotinib in both subgroups with and without brain metastases at treatment start (Supplementary Table 7).
Discussion
This study was designed to provide comparative efficacy evidence for a new medicine in a rare biomarker-defined lung cancer, ROS1+ NSCLC. The pivotal study was a single-arm trial with no comparative evidence since a comparative clinical trial of entrectinib versus crizotinib could potentially take many years to complete due to long recruitment periods and small numbers of patients. Using an external data source such as the FH database coupled with statistical modeling techniques, a comparator cohort was developed. In this study, we used TTD as the primary efficacy end point, and PFS and OS as secondary efficacy end points. The decision to use TTD as the primary end point was based on the availability of common variables between the trial and RWD cohorts.
The study sample sizes were small and not prespecified to determine superiority or non-inferiority in efficacy between arms. However, point estimates of treatment effectiveness showed benefit with entrectinib treatment in both primary and exploratory analyses. We do note that no formal statistical testing was planned and the primary analysis did not reach nominal significance level. A post-hoc power analysis using the final sample size (weighted) showed that the study would have been underpowered to detect a statistically significant effect in TTD (post-hoc power 37.2%), due to the effect estimate observed and the sample size obtained. Given the larger difference observed for PFS and OS, 80% power was attained for these outcomes.
This comparative effectiveness analysis between patients with ROS1+ NSCLC treated with entrectinib in clinical trials and a similar cohort treated with crizotinib in routine practice highlighted a number of key differences in clinical outcomes between the two populations. Entrectinib was associated with a longer median TTD compared with crizotinib (12.9 vs 8.2 months). Our primary end point, TTD, is a composite outcome evaluating progression together with the patient and clinician's judgment of tolerability, safety, and efficacy of the therapy. TTD was previously identified as a pragmatic end point for use in retrospective real-world evidence studies [18]. We acknowledge that rwP and clinical trial progression are inherently different. In addition to likely differences in scan frequency, objective and blinded progression assessments are not performed in the real world as they are in a clinical trial setting; rwP is based on clinician interpretation rather than objective measurements of computed tomography scans with RECIST criteria. The performance of the rwP end point employed in this study has been shown to closely mirror the outcomes in published clinical trial populations in patients with NSCLC [19,20]. We used BICR to define progression in our study, but found the results very similar when evaluating PFS and TTD using clinician assessment of progression in the trial.
It is important to recognize that the real-world variable used to calculate rwPFS in the crizotinib cohort is different from RECIST-based progression used in the clinical trial-based cohort. In addition, the calculation is contingent on the frequency of imaging assessments, which is likely to be lower in real-world practice, and might therefore bias the results in that cohort toward longer PFS. With these caveats, the comparison showed longer median PFS in the entrectinib cohort (16.8 vs 8.2 months). Additionally, median PFS for crizotinib in our RWD (8.2 months) was shorter than that reported in clinical trials of crizotinib in ROS1+ NSCLC (15.9 months per BICR [23] and 19.3 months per investigator [24]). However, similarities in baseline characteristics were observed in the ROS1+ NSCLC RWD crizotinib cohort and reported clinical trial cohorts [18]. We believe this difference is not specific to the FH population. Two RWD studies of crizotinib in European and Asian patients with ROS1+ NSCLC reported similar median PFS to our study (range: 9.1–9.6 months) [17,25]. However, direct comparison of different datasets should be made cautiously as the populations have inherent differences arising from medical practice at sites, biomarker testing, and population of patients captured. Two RWD studies in Chinese patients also reported a median PFS of 11.0–13.6 months, lower than the crizotinib clinical trial data [17,26], likely due to the demographics of the Chinese RWD populations resembling the clinical trial populations more closely than the FH population. For these reasons, we believe that our progression analyses should be viewed as exploratory.
Although OS is considered a relevant end point for comparative efficacy studies, the clinical trial data for this secondary end point were immature at the time of this analysis (median follow-up: 19.3 months) and median OS was not estimable. Nevertheless, the median OS observed in the crizotinib RWD cohort of 18.5 months (95% CI: 15.1–47.2) also suggests a possible benefit of entrectinib over crizotinib for this outcome, but confirmation of results is needed.
As the study includes a comparison with a non-randomized sample selected from a different population pool than the clinical trial, there were notable differences in recruitment criteria and baseline characteristics of patients from both cohorts that may have driven the differences in PFS observed, and limits valid cross-trial interpretations if minimal indirect matching is not implemented. We used several approaches to match the cohorts, including applying trial exclusion/inclusion criteria to the RWD cohort as a first step, and using PrS to balance any differences in cohorts before efficacy comparisons. PrS modeling was conducted to harmonize the two samples and allowed for a fairer comparison of treatment effects independently of selected patient baseline characteristics. This methodology is an accepted approach to reduce bias and estimate treatment effects across groups [22].
After weighting via a set of a priori-selected prognostic factors, an achievable balance in baseline covariates was obtained using SMDs and diagnostic assessments. This suggested IPTW was successful in creating a sample in which selected baseline covariates were similar between the entrectinib and crizotinib cohorts. While residual confounding may remain, the most critical covariates potentially associated to major confounding were likely included in this analysis, since the measured confounders may also serve as proxies for unmeasured factors (such as general state of health). Additionally, sensitivity analyses conducted with differing model specifications for outcome assessment and population selection did not modify the results or conclusions. It is of particular relevance that both sensitivity analyses used on restricted populations limit heterogeneity between cohorts and strengthen the HR away from no effect.
Our results suggest that both patients with and without brain metastases at baseline benefit from entrectinib. After adjusting for various prognostic factors, we found a similar benefit with entrectinib versus crizotinib, based on CNS status at baseline. Assessments from the clinic and clinical trials of the presence of brain metastases and their progression are likely different and could bias estimates.
Other limitations such as reasons for treatment discontinuation that could not be determined for the crizotinib cohort and data quality should also be noted. Although the RWD were obtained under strict quality control methods [19,27], inherent limitations of such data exist, for example, the potential for missing information due to documentation practices or partial receipt of care outside of the FH network. Finally, the potential impact of missing data on our findings should be noted, as only 0.3% (134/48,935) of patients in the crizotinib cohort had a ROS1 fusion, which is lower than the estimated prevalence of 1–2% reported for this fusion. However, it is likely that this percentage may reflect a lack of testing for this marker in the real-world setting, consistent with reports showing lack of testing for more well established EGFR and ALK biomarkers in NSCLC [28].
Conclusion
This analysis of clinical trial and real-world cohorts suggests that entrectinib treatment in clinical trials may be associated with longer TTD and PFS in patients with ROS1+ NSCLC compared with a matched real-world crizotinib population of patients treated with crizotinib in routine clinical practice. The use of RWD has provided an alternative approach in generating comparative efficacy evidence for licensing of new drugs, in a setting where a prospective randomized trial would not be feasible to conduct, thereby accelerating the development of new molecular entities.
•
Given the rarity of ROS1 gene fusions, there has been no direct head-to-head study comparing the efficacy of entrectinib with crizotinib in patients with ROS1 fusion-positive (ROS1+) NSCLC.
•
We conducted a comparative effectiveness analysis between entrectinib-treated adults with ROS1+ NSCLC enrolled in Phase I/II clinical trials (n = 94) and crizotinib-treated adults with ROS1+ NSCLC identified from a real-world database (n = 65).
•
Trial eligibility criteria were applied to the crizotinib cohort and propensity score modelling was used to balance any between-cohort differences.
•
Outcomes assessed included time-to-treatment discontinuation (TTD; primary objective), OS (secondary objective) and PFS (exploratory objective).
•
Entrectinib was associated with a longer median TTD and PFS than crizotinib, but OS data for entrectinib were immature and median OS was not estimable.
•
After adjusting for prognostic factors, a similar benefit was evident with entrectinib versus crizotinib in secondary efficacy analyses based on CNS status at baseline.
•
Results of this analysis suggest that entrectinib administered to patients with ROS1+ NSCLC in clinical trials may be associated with longer TTD than a matched real-world population treated with crizotinib.
•
Real-world data provide an alternative means to generate comparative efficacy evidence in a setting where a prospective randomized trial may not be feasible.
Author contributions
G Crane, RC Doebele, MG Krebs and NJ Meropol contributed to conception and design. Patient recruitment was carried out by RC Doebele. The principal investigators at contributing sites were RC Doebele and MG Krebs. G Crane, H Trinh, M Martinec and MG Krebs analyzed the data. All authors were involved in data interpretation, writing of the report and approval of the report.
Acknowledgments
RC Doebele had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. The authors would like to thank the patients treated with crizotinib whose real-world data contributed to this study. The authors would also like to thank the patients, their families and the participating study centers from the entrectinib clinical trials. MG Krebs would like to acknowledge support by NIHR Manchester Biomedical Research Centre, NIHR Manchester Clinical Research Facility at The Christie and Manchester Experimental Cancer Medicine Centre, Manchester, UK.
In memory of our dear colleague and co-author T Riehl.
Financial & competing interests disclosure
This study was funded and conducted by F Hoffmann-La Roche Ltd. RC Doebele declares consulting fees from Ignyta, Genentech, Inc./F. Hoffmann-La Roche Ltd, AstraZeneca, Anchiano and Rain Therapeutics; royalties or licensing fees for intellectual property from Ignyta, Abbott Molecular, Genentech, Inc./F. Hoffmann-La Roche Ltd, Foundation Medicine, Black Diamond, Pearl River, Voronoi, Takeda, Scorpion and Rain Therapeutics; stock ownership in Rain Therapeutics; is an employee of Rain Therapeutics. MG Krebs declares honoraria from F Hoffmann-La Roche Ltd; consulting/advisory role for F Hoffmann-La Roche Ltd, Achilles Therapeutics, Octimet, Janssen, Bayer, Seattle Genetics; research funding from F Hoffmann-La Roche Ltd; BerGenBio; travel/accommodation/expenses from AstraZeneca, BerGenBio. R Martina declares consulting/advisory role for F Hoffmann-La Roche Ltd, Dompe Pharmaceuticals. H Trinh, T Riehl and W Wong declare stock and ownership interests and are employees of Genentech, Inc. (a member of the Roche Group). L Perez, G Crane and M Martinec declare stock and ownership interests and are employees of F. Hoffmann-La Roche Ltd. NJ Meropol is an employee of Flatiron Health, Inc., an independent subsidiary of the Roche Group, and reports equity interests in Flatiron Health and F Hoffmann-La Roche Ltd. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Third-party medical writing assistance, under the direction of the authors, was provided by L Carrier and C Kennerley of Ashfield MedComms, an Ashfield Health company, and was funded by F Hoffmann-La Roche Ltd.
Ethical conduct of research
All studies reported in this secondary analysis were conducted in accordance with the principles of the Declaration of Helsinki and Good Clinical Practice guidelines and all patients provided written informed consent. Protocols were approved by the relevant institutional review boards and/or ethics committees.
Data sharing statement
The data were generated and analyzed under the auspices of Roche, which is a member of the Vivli Center for global clinical research data (https://vivli.org/ourmember/roche/). Roche will share/allow access to individual patient-level data from the clinical trials via Vivli, providing certain criteria are met. Please see the criteria and exceptions on the Roche member section of the Vivli homepage here. Please see also the Roche Global Data Sharing Policy for more details. To request access to individual patient-level data from the clinical trials, first locate the clinical trial in Vivli (https://search.vivli.org/ requires sign up and log in) using the trial registration number (given above), then click the ‘Request Study’ button and follow the instructions. In the event that you cannot see a specific study in the Roche list, an Enquiry Form can be submitted to confirm the availability of the specific study. To request access to related clinical study documents (e.g,: protocols, CSR, safety reports), please use Roche's Clinical study documents request form: https://www.roche.com/research_and_development/who_we_are_how_we_work/research_and_clinical_trials/our_commitment_to_data_sharing/clinical_study_documents_request_form.htm
Open access
This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
Supplementary Material
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
1.
Chen Z, Fillmore CM, Hammerman PS, Kim CF, Wong KK. Non-small-cell lung cancers: a heterogeneous set of diseases. Nat. Rev. Cancer 14(8), 535–546 (2014).
2.
Bendaly E, Dalal AA, Culver K et al. Monitoring for and characterizing crizotinib progression: a chart review of ALK-positive non-small cell lung cancer patients. Adv. Ther. 34(7), 1673–1685 (2017).
3.
Sehgal K, Patell R, Rangachari D, Costa DB. Targeting ROS1 rearrangements in non-small cell lung cancer with crizotinib and other kinase inhibitors. Transl. Cancer Res. 7(Suppl. 7), S779–S786 (2018).
• Overview of clinical trials of targeted therapies for ROS1 fusion-positive non-small-cell lung cancer (NSCLC) and discussion of important issues in the clinical management of this patient group.
4.
Davies KD, Doebele RC. Molecular pathways: ROS1 fusion proteins in cancer. Clin. Cancer Res. 19(15), 4040–4045 (2013).
5.
Bergethon K, Shaw AT, Ou SH et al. ROS1 rearrangements define a unique molecular class of lung cancers. J. Clin. Oncol. 30(8), 863–870 (2012).
6.
Dugay F, Llamas-Gutierrez F, Gournay M et al. Clinicopathological characteristics of ROS1- and RET-rearranged NSCLC in Caucasian patients: data from a cohort of 713 non-squamous NSCLC lacking KRAS/EGFR/HER2/BRAF/PIK3CA/ALK alterations. Oncotarget 8(32), 53336–53351 (2017).
7.
Davies KD, Le AT, Theodoro MF et al. Identifying and targeting ROS1 gene fusions in non-small cell lung cancer. Clin. Cancer Res. 18(17), 4570–4579 (2012).
8.
Drilon A, Siena S, Ou SI et al. Safety and antitumor activity of the multitargeted pan-TRK, ROS1, and ALK inhibitor entrectinib: combined results from two Phase I trials (ALKA-372-001 and STARTRK-1). Cancer Discov. 7(4), 400–409 (2017).
9.
Rolfo C, Ruiz R, Giovannetti E et al. Entrectinib: a potent new TRK, ROS1, and ALK inhibitor. Expert Opin. Investig. Drugs 24(11), 1493–1500 (2015).
10.
Ardini E, Menichincheri M, Banfi P et al. Entrectinib, a pan-TRK, ROS1, and ALK inhibitor with activity in multiple molecularly defined cancer indications. Mol. Cancer Ther. 15(4), 628–639 (2016).
11.
Doebele R, Ahn M, Siena S et al. OA02.01 Efficacy and safety of entrectinib in locally advanced or metastatic ROS1 fusion-positive non-small cell lung cancer (NSCLC). J. Thorac. Oncol. 13(10), S321–S322 (2018).
12.
Doebele RC, Drilon A, Paz-Ares L et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: integrated analysis of three Phase I–2 trials. Lancet Oncol. 21(2), 271–282 (2020).
•• Entrectinib was well tolerated and demonstrated robust antitumor activity in patients with solid tumors harboring NTRK1/2/3 gene fusions, including those with CNS disease.
13.
Drilon A, Siena S, Dziadziuszko R et al. Entrectinib in ROS1 fusion-positive non-small-cell lung cancer: integrated analysis of three Phase I–2 trials. Lancet Oncol. 21(2), 261–270 (2020).
•• Entrectinib showed durable disease control (objective response rate [ORR] 77.4%; median PFS 19.0 months; median DoR 24.6 months) and was well tolerated in an integrated analysis of clinical trial data from patients with ROS1 fusion-positive NSCLC.
14.
Berger ML, Curtis MD, Smith G, Harnett J, Abernethy AP. Opportunities and challenges in leveraging electronic health record data in oncology. Future Oncol. 12(10), 1261–1274 (2016).
15.
Abernethy AP, Gippetti J, Parulkar R, Revol C. Use of electronic health record data for quality reporting. J. Oncol. Pract. 13(8), 530–534 (2017).
16.
Metro G, Tazza M, Matocci R, Chiari R, Crino L. Optimal management of ALK-positive NSCLC progressing on crizotinib. Lung Cancer 106, 58–66 (2017).
17.
Liu C, Yu H, Chang J et al. Crizotinib in chinese patients with ROS1-rearranged advanced non small-cell lung cancer in routine clinical practice. Target Oncol. 14(3), 315–323 (2019).
18.
Gong Y, Kehl KL, Oxnard GR, Khozin S, Mishra-Kalyani PS, Blumenthal GM. Time to treatment discontinuation (TTD) as a pragmatic endpoint in metastatic non-small cell lung cancer (mNSCLC): a pooled analysis of 8 trials. J. Clin. Oncol. 36(Suppl. 15), 9064–9064 (2018).
19.
Griffith SD, Tucker M, Bowser B et al. Generating real-world tumor burden endpoints from electronic health record data: comparison of RECIST, radiology-anchored, and clinician-anchored approaches for abstracting real-world progression in non-small cell lung cancer. Adv. Ther. 36(8), 2122–2136 (2019).
20.
Griffith SD, Miksad RA, Calkins G et al. Characterizing the feasibility and performance of real-world tumor progression end points and their association with overall survival in a large advanced non-small-cell lung cancer data set. JCO Clin. Cancer Inform. 3, 1–13 (2019).
• Retrospective characterization of real-world progression data from Flatiron Health's electronic health record-derived database, showing it to be reliable and clinically relevant in the assessment of advanced NSCLC.
21.
Curtis MD, Griffith SD, Tucker M et al. Development and validation of a high-quality composite real-world mortality endpoint. Health Serv. Res. 53(6), 4460–4476 (2018).
22.
Austin PC, Stuart EA. Moving towards best practice when using inverse probability of treatment weighting (IPTW) using the propensity score to estimate causal treatment effects in observational studies. Stat. Med. 34(28), 3661–3679 (2015).
23.
Wu YL, Yang JC, Kim DW et al. Phase II study of crizotinib in East Asian patients with ROS1-positive advanced non-small-cell lung cancer. J. Clin. Oncol. 36(14), 1405–1411 (2018).
•• Crizotinib demonstrated clinically meaningful benefit (ORR 71.7%; median PFS 15.9 months) and durable responses (median DoR 19.7 months) in a Phase II study in East Asian patients with ROS1-positive advanced NSCLC.
24.
Shaw AT, Riely GJ, Bang YJ et al. Crizotinib in ROS1-rearranged advanced non-small-cell lung cancer (NSCLC): updated results, including overall survival, from PROFILE 1001. Ann. Oncol. 30(7), 1121–1126 (2019).
•• Crizotinib showed marked antitumor activity (ORR 72.0%; median PFS 19.2 months; median DoR 17.6 months) in patients with advanced ROS1-positive NSCLC in the global Phase I PROFILE 1001 study.
25.
Mazieres J, Zalcman G, Crino L et al. Crizotinib therapy for advanced lung adenocarcinoma and a ROS1 rearrangement: results from the EUROS1 cohort. J. Clin. Oncol. 33(9), 992–999 (2015).
26.
Zeng L, Li Y, Xiao L et al. Crizotinib presented with promising efficacy but for concomitant mutation in next-generation sequencing-identified ROS1-rearranged non-small-cell lung cancer. Onco Targets Ther. 11, 6937–6945 (2018).
27.
Khozin S, Abernethy AP, Nussbaum NC et al. Characteristics of real-world metastatic non-small cell lung cancer patients treated with nivolumab and pembrolizumab during the year following approval. Oncologist 23(3), 328–336 (2018).
28.
Ruggiero JE, Rughani J, Neiman J et al. Real-world concordance of clinical practice with ASCO and NCCN guidelines for EGFR/ALK testing in aNSCLC. J. Clin. Oncol. 35(Suppl. 8), 212–212 (2017).
Information & Authors
Information
Published In
Pages: 1271 - 1282
PubMed: 34427452
Copyright
© 2021 Laura Perez. This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 Unported License
History
Received: 4 June 2021
Accepted: 9 August 2021
Published online: 24 August 2021
Keywords:
Topics
Authors
Funding Information
F Hoffmann-La Roche Ltd
Metrics & Citations
Metrics
Article Usage
Article usage data only available from February 2023. Historical article usage data, showing the number of article downloads, is available upon request.
Citations
How to Cite
Comparative effectiveness analysis between entrectinib clinical trial and crizotinib real-world data in ROS1+ NSCLC. (2021) Journal of Comparative Effectiveness Research. DOI: 10.2217/cer-2021-0131
Export citation
Select the citation format you wish to export for this article or chapter.
Citing Literature
- Yi Piao, Yuya Kimijima, Michihiko Nakamura, Nobutomo Matsui, Characteristics and Future Perspectives of Available Electronic Health Record Databases in Japan, Drugs - Real World Outcomes, 10.1007/s40801-026-00562-w, (2026).
- Jeroen P. Jansen, Michael P. Douglas, Kathryn A. Phillips, Combining Simulation Model-Based Outcomes With County-Level Data for Geographic Health Equity Impact Evaluations of New Interventions, Value in Health, 10.1016/j.jval.2025.07.025, 29, 1, (34-45), (2026).
- Hiroshi Tanaka, The Third Revolution of Medicine by Big data and Artificial Intelligence, Chem-Bio Informatics Journal, 10.1273/cbij.25.53, 25, 0, (53-70), (2025).
- Ahan Bhatt, Nang Yone, Mumtu Lalla, Hyein Jeon, Haiying Cheng, Molecular Diagnosis, Clinical Trial Representation, and Precision Medicine in Minority Patients with Oncogene-Driven Lung Cancer, Cancers, 10.3390/cancers17121950, 17, 12, (1950), (2025).
- Takashi Seto, Sayuri Nakane, Lyu Ji, Yuya Ueda, Hiroshi Sugano, Nobuki Takei, Mizuki Kobayashi, Ayako Murayama, Noboru Yamamoto, Real-world safety and effectiveness of entrectinib in Japanese patients with ROS1 gene fusion-positive, unresectable, advanced/recurrent non-small cell lung cancer: Post-marketing surveillance, Lung Cancer, 10.1016/j.lungcan.2025.108478, 203, (108478), (2025).
- Lishi Lin, Merel J.J. Lucassen, Vincent van der Noort, Toine C.G. Egberts, Jos H. Beijnen, Alwin D.R. Huitema, The feasibility of using real world data as external control arms in oncology trials, Drug Discovery Today, 10.1016/j.drudis.2025.104324, 30, 3, (104324), (2025).
- Monica Daigl, Seye Abogunrin, Felipe Castro, Sarah F McGough, Rachele Hendricks Sturrup, Cornelis Boersma, Keith R Abrams, Advancing the role of real-world evidence in comparative effectiveness research, Journal of Comparative Effectiveness Research, 10.57264/cer-2024-0101, 13, 12, (2024).
- Ernest Nadal, Nada Rifi, Sarah Kane, Sokhna Mbacke, Lindsey Starkman, Beatrice Suero, Hannah Le, Imtiaz A. Samjoo, Efficacy and safety of crizotinib in the treatment of advanced non-small cell lung cancer with ROS1 gene fusion: a systematic literature review and meta-analysis of real-world evidence, Lung Cancer, 10.1016/j.lungcan.2024.107816, 192, (107816), (2024).
- Francesco Perri, Roberta Fusco, Francesco Sabbatino, Morena Fasano, Alessandro Ottaiano, Marco Cascella, Maria Luisa Marciano, Monica Pontone, Giovanni Salzano, Maria Elena Maiello, Massimo Montano, Ester Calogero, Roberta D’Aniello, Piera Maiolino, Fortunato Ciardiello, Alessia Zotta, Salvatore Alfieri, Franco Ionna, Translational Insights in the Landscape of Salivary Gland Cancers: Ready for a New Era?, Cancers, 10.3390/cancers16050970, 16, 5, (970), (2024).
- Yumi Yasui, Masataka Matsumoto, Teruaki Hyakudo, Masahiko Nishii, Saki Ito, Masahiro Katsurada, Yuko Kono, Kiyonobu Takatsuki, Yoshihiro Nishimura, A Case of Successful Treatment of ROS1-positive Granulocyte Colony-stimulating Factor-producing Lung Adenocarcinoma with an ROS1 Tyrosine Kinase Inhibitor, Haigan, 10.2482/haigan.64.45, 64, 1, (45-49), (2024).