Estimating the health benefits of timely diagnosis and treatment of transthyretin amyloid cardiomyopathy
Publication: Journal of Comparative Effectiveness Research
Abstract
Aim: Delayed diagnosis of transthyretin amyloid cardiomyopathy (ATTR-CM) represents a missed opportunity for intervention. This study estimates the health benefits of timely diagnosis and treatment with tafamidis. Methods: A disease simulation model was developed to predict health outcomes under scenarios of timely and delayed diagnosis and treatment. Efficacy and quality of life (QoL) profiles were derived from the pivotal tafamidis trial and diagnostic delay durations from the literature. Results: Timely diagnosis and treatment were predicted to extend mean life expectancy by 5.46 and 7.76 years, relative to delayed diagnosis, for wild-type and hereditary ATTR-CM, respectively. Corresponding QALY gains were 4.50 and 6.22. Conclusion: Timely diagnosis and treatment with tafamidis are predicted to significantly improve survival and QoL for ATTR-CM patients.
Introduction
Transthyretin amyloid cardiomyopathy (ATTR-CM) is a progressive and ultimately fatal disease caused by the accumulation of amyloid fibrils in the heart muscle [1,2]. This leads to restrictive cardiomyopathy, progressive heart failure and increased risk of conduction abnormalities and arrhythmias [1]. The more common wild-type form (ATTRwt-CM) is associated with aging, with symptom onset typically occurring at age 60 years or older, and is diagnosed predominantly in males [3–5]. The prevalence of ATTRwt-CM is not well characterized, but the condition is thought to be under recognized [1,2]. Hereditary ATTR-CM, also known as variant (ATTRv-CM), is caused by autosomal dominant mutations in the TTR gene [6]. ATTRv-CM is rarer and occurs in geographical and ethnic clusters [6]. In the USA, the most common mutation, Val122Ile (also reported as p.Val142Ile), is almost exclusively identified in the African–American population, where it has a prevalence of approximately 3–4% [7,8].
The prognosis for ATTR-CM is poor, with typical survival from diagnosis of 2–6 years [2]. As the disease progresses, patients experience worsening symptoms of heart failure, including fatigue, dyspnea (shortness of breath), reduced exercise capacity and impaired health-related quality of life (HRQoL) [2,9,10].
Diagnostic delay is common [6,11–13], with patients often not correctly diagnosed for several years after symptom onset. The reasons for this include low awareness of ATTR-CM among clinicians [6,11,14], symptom overlap with other more common causes of heart failure and the historical need for invasive diagnosis via cardiac biopsy [15,16]. In patients with mixed phenotype ATTRv-CM, diagnosis may also be complicated by the presence of neurological manifestations [2,17], with a multidisciplinary approach and close communication between cardiologists and neurologists often necessary. Since 2016, a range of recommendations on noninvasive diagnostic techniques have been published [2,15,18,19], as have suggested diagnostic ‘red flags’ for ATTR-CM [2,6]. Together, these have increased awareness and made diagnosis easier, although uptake outside of specialist centers remains inconsistent [20,21].
Another important contributing factor to diagnostic delay has been the absence of a disease-modifying treatment, which together with the need for invasive diagnosis, previously limited the incentive to refer patients for specialist investigation. This situation changed with the introduction of tafamidis, which was approved for the treatment of ATTR-CM by the US FDA in 2019 and by the EMA in 2020 [22,23]. In the pivotal Phase III trial (tafamidis in transthyretin cardiomyopathy clinical trial; ATTR-ACT), treatment with tafamidis reduced all-cause mortality compared with a placebo (HR: 0.70 [95% CI: 0.51–0.95], p = 0.0259) [24]. Tafamidis was also associated with favorable effects on HRQoL and functional capacity [24].
With a disease-modifying treatment now available, diagnostic delay represents a missed opportunity for intervention to extend and improve life for patients with ATTR-CM through treatment. Furthermore, delayed diagnosis is likely to result in more advanced disease at treatment initiation than would otherwise be the case [25,26], reducing the potential to benefit from treatment. The objective of this study was to estimate the expected health benefits of timely diagnosis and treatment of ATTR-CM with tafamidis in the US setting, using a disease simulation model.
Methods
A disease simulation model was developed to characterize the natural history of disease for individuals with ATTR-CM and to predict long-term health outcomes under scenarios of timely and delayed diagnosis followed by treatment with tafamidis.
Model conceptualization
Model conceptualization was informed by relevant existing literature, including publicly available health technology assessment dossiers for chronic heart failure therapies (e.g., ivabradine [27] and sacubitril valsartan [28]). A panel of clinical experts also contributed to the model's design to ensure face validity and that the model provided an accurate approximation of clinical practice.
Model overview
A discrete-time, cohort-level Markov state-transition model was developed to simulate the chronic burden of ATTR-CM over a lifetime horizon.
To capture the natural disease progression of ATTR-CM and the patient experience [29], model health states were characterized by the New York Heart Association (NYHA) functional classification of heart failure (NYHA classes I–IV). This widely used classification system is based on the degree to which physical activity is limited and the activity thresholds resulting in heart failure symptoms [29]. NYHA class is also a statistically significant predictor of both HRQoL and survival [30–32].
For a defined cohort of patients, disease progression, characterized by transitions through the NYHA classes, is simulated until death due to general or disease-related causes. Consistent with clinical expectations and in line with trial outcomes [33], the risk of death due to disease-related causes is conditional upon NYHA class, with patients with more advanced disease (i.e., higher NYHA class) subject to greater risk. The rate at which patients progress through NYHA classes in the model is governed by transition probabilities; these describe the probability that patients will move from one class to another over a given period of time. HRQoL is characterized by utility weights assigned to each NYHA class.
A schematic diagram of the model structure is presented in Figure 1; technical specification of the model implementation is provided in the Supplementary material (See S2.1).
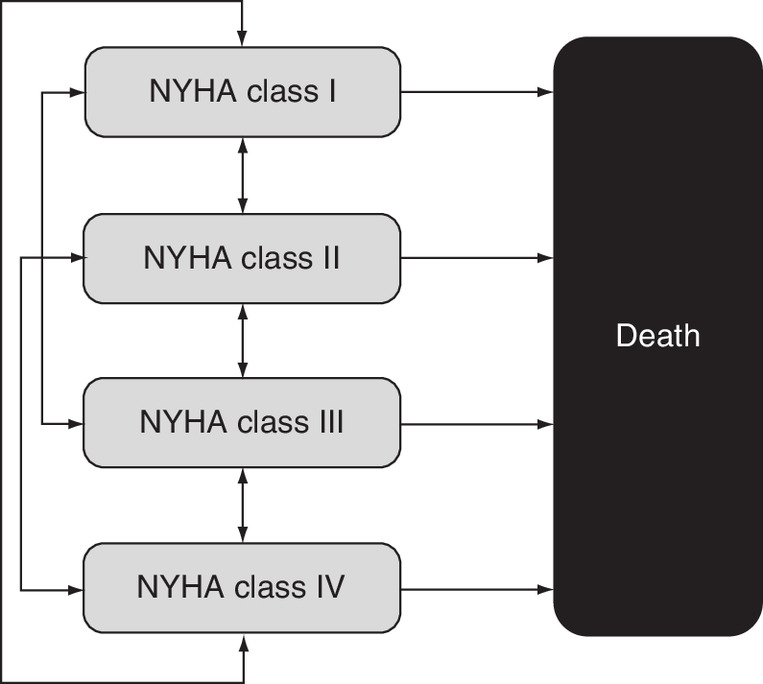
Figure 1. Schematic diagram of the structure of the model.
NYHA: New York Heart Association.
Model application: primary analyses
The model was configured to predict outcomes for cohorts of patients under scenarios in which diagnosis and treatment with tafamidis were timely or delayed, with timely diagnosis and treatment defined by younger age and less severe disease (i.e., lower NYHA class) at initiation.
At model initiation, simulated patients were assumed to be in NYHA class I or II, with the applied distribution derived from the number of patients in each of these classes at baseline in ATTR-ACT [33]. Diagnostic delay durations and baseline demographic characteristics were informed by the findings of a targeted literature review characterizing the extent and consequences of delayed diagnosis and misdiagnosis for patients with ATTR-CM; the review identified 22 studies reporting on time from symptom onset to diagnosis in patients with ATTR-CM [13]. The clinical plausibility of the identified diagnostic delay durations and the representativeness of modeled patient demographics were verified by a panel of clinical experts. Independent analyses were conducted for ATTRwt-CM and ATTRv-CM patients to reflect the difference in diagnostic delay and demographic profiles seen between the two forms in the literature [13], and in line with clinical expectations. Treatment with tafamidis was assumed to commence at diagnosis.
Treatment efficacy, captured via the estimation of survival and NYHA class status, was based on analysis of individual patient-level data from ATTR-ACT and its LTE (preliminary August 2019 database lock) [24,33,34]. While on treatment, simulated patients assume efficacy profiles derived from the pooled tafamidis meglumine 20 and 80 mg dosing arms of ATTR-ACT and its LTE. Prior to commencing treatment (i.e., during the diagnostic delay period), patients assume efficacy profiles derived from ATTR-ACT's placebo arm. US preference weights [36] were used to derive treatment-specific NYHA class utility values from EuroQoL five-dimension 3-level (EQ-5D-3L) data collected directly from patients during the trial.
The expected benefits of timely diagnosis and treatment were quantified in terms of incremental gains in life expectancy and quality-adjusted life years (QALYs). A schematic diagram illustrating the modeling framework for a 5-year delay is presented in Figure 2. Baseline patient demographic, baseline NYHA class and delay duration profiles applied in the primary analysis are presented in Table 1, and a descriptive summary of all other modeled input profiles is provided in Table 2; further detail on the applied input profiles is provided in the Supplementary material (see S2.2 and S2.3).
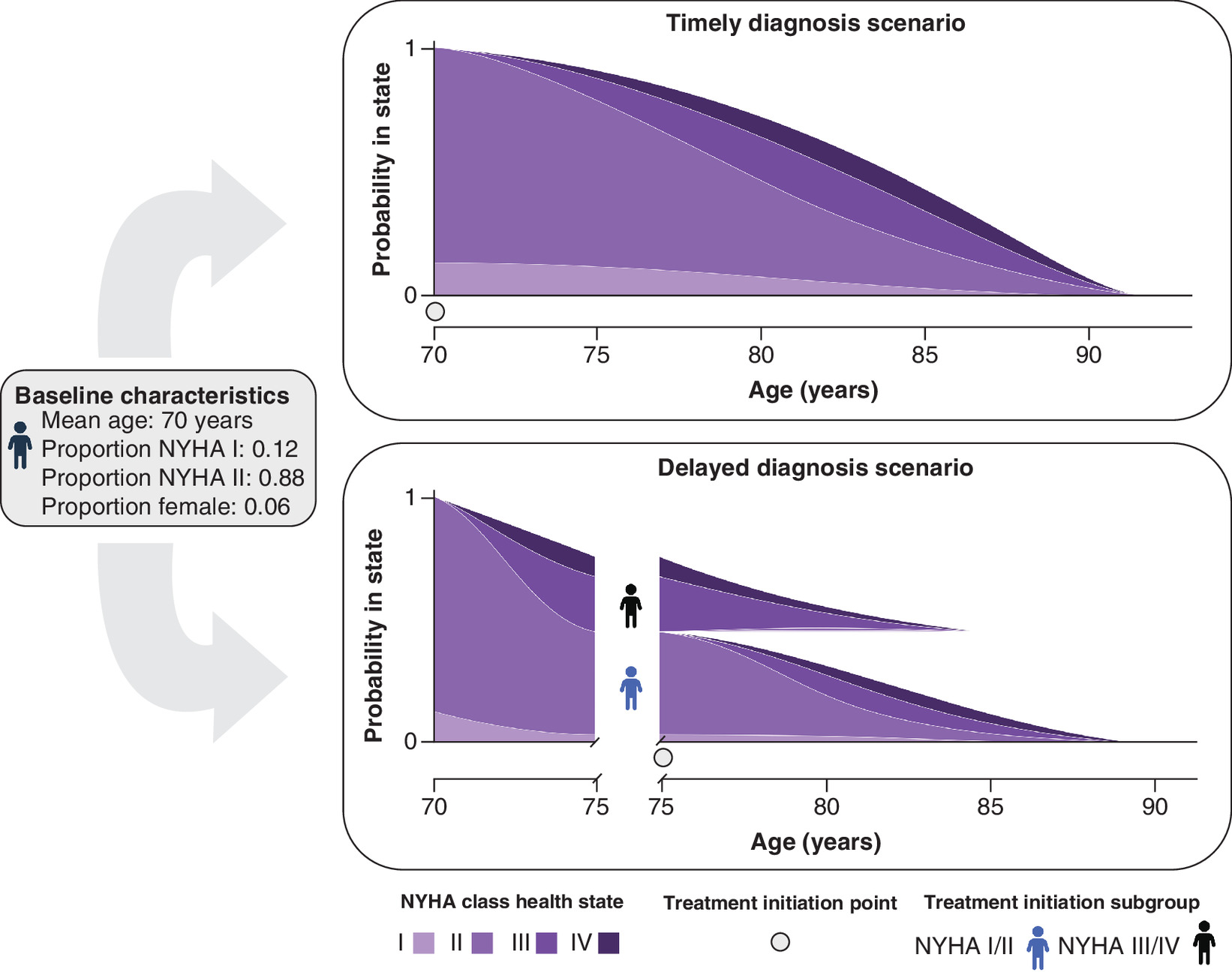
Figure 2. Schematic illustrating the applied modelling framework for a 5-year diagnostic delay.
NYHA: New York Heart Association.
| Parameter | Value | Source | Ref. |
|---|---|---|---|
| ATTRwt-CM | |||
| Mean age at baseline (years) | 70 | Rozenbaum et al.† | [13] |
| Proportion female | 0.06 | ||
| Baseline NYHA class distribution | |||
| Proportion NYHA I | 0.12 | ATTR-ACT ATTRwt-CM population | [33] |
| Proportion NYHA II | 0.88 | ||
| Mean delay duration (years) | 6.08 (min: 1.25; max: 7.17) | Rozenbaum et al.‡ | [13] |
| ATTRv-CM | |||
| Mean age at baseline (years) | 60 | Rozenbaum et al.† | [13] |
| Proportion female | 0.27 | ||
| Baseline NYHA class distribution | |||
| Proportion NYHA I | 0.12 | ATTR-ACT ATTRv-CM population | [33] |
| Proportion NYHA II | 0.88 | ||
| Mean delay duration (years) | 5.67 (min: 1.75; max: 6.50) | Rozenbaum et al.‡ | [13] |
†
Baseline age and proportion female values were derived from the studies employed to inform delay durations [13]; reported or derived mean age at onset was employed to inform baseline age, with values rounded to the nearest 5-year multiple.
‡
To align with the model cycle length, reported diagnostic delay durations were rounded down to the nearest month.
ATTR-CM: Transthyretin amyloid cardiomyopathy; ATTRv: Variant ATTR amyloidosis; ATTRwt: Wild-type ATTR amyloidosis; NYHA: New York Heart Association.
| Parameter group | Parameter | Description | Source | Ref. |
|---|---|---|---|---|
| Survival | Background (general population) mortality | US age- and sex-specific national life tables | Arias et al. (CDC) | [35] |
| Disease-related mortality | Treatment-specific survival models | ATTR-ACT & LTE | [33,34] | |
| NYHA class mortality-relative risks† | Treatment specific NYHA class relative risks of mortality | ATTR-ACT & LTE | [33,34] | |
| Disease progression | NYHA class transition probabilities | Treatment-specific NYHA class transition matrices | ATTR-ACT & LTE | [33,34] |
| Health-related quality of life | NYHA class health state utilities | Treatment and NYHA class specific utility values | ATTR-ACT; Shaw et al. | [33,36] |
†
Employed to inform NYHA class-specific disease-related mortality risk, only.
CV: Cardiovascular; NYHA: New York Heart Association.
Model application: sensitivity & scenario analyses
Analyses were carried out to assess the sensitivity of model predictions to changes in the baseline age (±5 years) and the sex distribution of the cohort (proportion female ±20%). In addition, several scenario analyses were undertaken to evaluate the impact of key assumptions on modeled outcomes. The survival models used to characterize disease-related mortality in the primary analyses were chosen as they best represent the observed trial data and have underlying profiles consistent with clinical expectations. However, in line with best practice, scenario analyses evaluated the impact of employing alternative survival models to characterize disease-related mortality (see S2.3 for details). Patient NYHA class status was assessed at a frequency of 6 months in ATTR-ACT and its LTE. In the primary analyses, transition matrices governing movement between NYHA classes in the model were informed by transitions observed across all NYHA class assessment intervals. This approach was adopted to ensure optimal use of the data points (see S2.2 for details). However, progression rates for the early within-trial assessment intervals (months 0–6 and 6–12) showed some differences to those later in the trial. The impact of only modeling transitions observed over each of these 6-month intervals for these early periods was assessed via scenario analysis. Finally, a scenario evaluated outcomes based on efficacy profiles derived from the tafamidis meglumine 80 mg dosing arm alone. The primary analysis of ATTR-ACT compared the pooled tafamidis arms (tafamidis meglumine 20 and 80 mg dosing) with the placebo arm [24], but the FDA- and EMA-recommended doses are tafamidis meglumine 80 mg and the 61 mg free acid bioequivalent dose, respectively [22,23]. Post hoc analysis of data from ATTR-ACT and its LTE confirmed that the 80 mg dose had a significantly greater survival benefit over placebo compared with the 20 mg dose, with no dose-related safety concerns [37].
Analytic software
The disease simulation model was developed in Microsoft Excel 2016® (Microsoft Corporation, WA, USA). R version 3.6.3 (R Foundation for Statistical Computing, Vienna, Austria) was used to conduct statistical analyses informing the model input profiles and for the production of results figures.
Results
Primary analysis
Per-patient health outcomes for the timely and delayed diagnosis and treatment scenarios modeled in the primary analyses are presented in Table 3. In ATTRwt-CM patients, timely diagnosis and treatment with tafamidis were predicted to extend mean life expectancy by 5.46 years relative to delayed diagnosis (mean delay: 6.08 years). For ATTRv-CM patients, timely diagnosis and treatment were predicted to result in life expectancy gains of 7.76 years relative to delayed diagnosis (mean delay: 5.67 years). Most of the benefit was accrued in the NYHA classes corresponding to less advanced heart failure (classes I and II). With timely diagnosis and treatment, ATTRwt-CM patients were predicted to spend approximately twice as long (4.14 additional years) in NYHA class I and II than if they had experienced the mean delay; ATTRv-CM patients were predicted to spend almost two and a half times longer (5.09 additional years) in these classes. Resultant QALY gains were predicted to be 4.50 and 6.22 for ATTRwt-CM and ATTRv-CM, respectively.
| ATTR-CM type | Scenario | Life years | QALYs | ||
|---|---|---|---|---|---|
| ATTRwt-CM | Timely diagnosis scenario | 11.65 | 9.31 | ||
| Delayed diagnosis scenario (6.08-year delay) | 6.19 | 4.81 | |||
| Incremental | 5.46 | 4.50 | |||
| ATTRv-CM | Timely diagnosis scenario | 12.62 | 10.01 | ||
| Delayed diagnosis scenario (5.67-year delay) | 4.86 | 3.80 | |||
| Incremental | 7.76 | 6.22 | |||
| Time spent in NYHA class (years)† | |||||
|---|---|---|---|---|---|
| NYHA I | NYHA II | NYHA III | NYHA IV | ||
| ATTRwt-CM | Timely diagnosis scenario | 1.31 | 7.01 | 3.14 | 0.20 |
| Delayed diagnosis scenario (6.08-year delay) | 0.53 | 3.64 | 1.84 | 0.18 | |
| Incremental | 0.78 | 3.36 | 1.30 | 0.02 | |
| ATTRv-CM | Timely diagnosis scenario | 1.69 | 6.89 | 3.60 | 0.43 |
| Delayed diagnosis scenario (5.67-year delay) | 0.48 | 3.01 | 1.21 | 0.16 | |
| Incremental | 1.21 | 3.88 | 2.40 | 0.27 | |
Apparent differences in the reported values are due to rounding.
†
The time spent in each NYHA class is determined by the baseline NYHA class distribution of the modeled patient cohort, and the modeled NYHA class transition and mortality risk profiles. The majority of the modeled patient cohort (88%) were in NYHA class II at baseline with the remainder (12%) in NYHA class I, for both ATTRwt-CM and ATTRv-CM.
ATTR-CM: Transthyretin amyloid cardiomyopathy; ATTRv: Variant ATTR amyloidosis; ATTRwt: Wild-type ATTR amyloidosis; Inc.: Incremental; NYHA: New York Heart Association; QALY: Quality-adjusted life year.
As expected, for both ATTRwt-CM (Figure 3) and ATTRv-CM (Figure 4) patients, predicted life expectancy and QALY gains associated with timely diagnosis and treatment were greater when compared with outcomes for more lengthy delays.
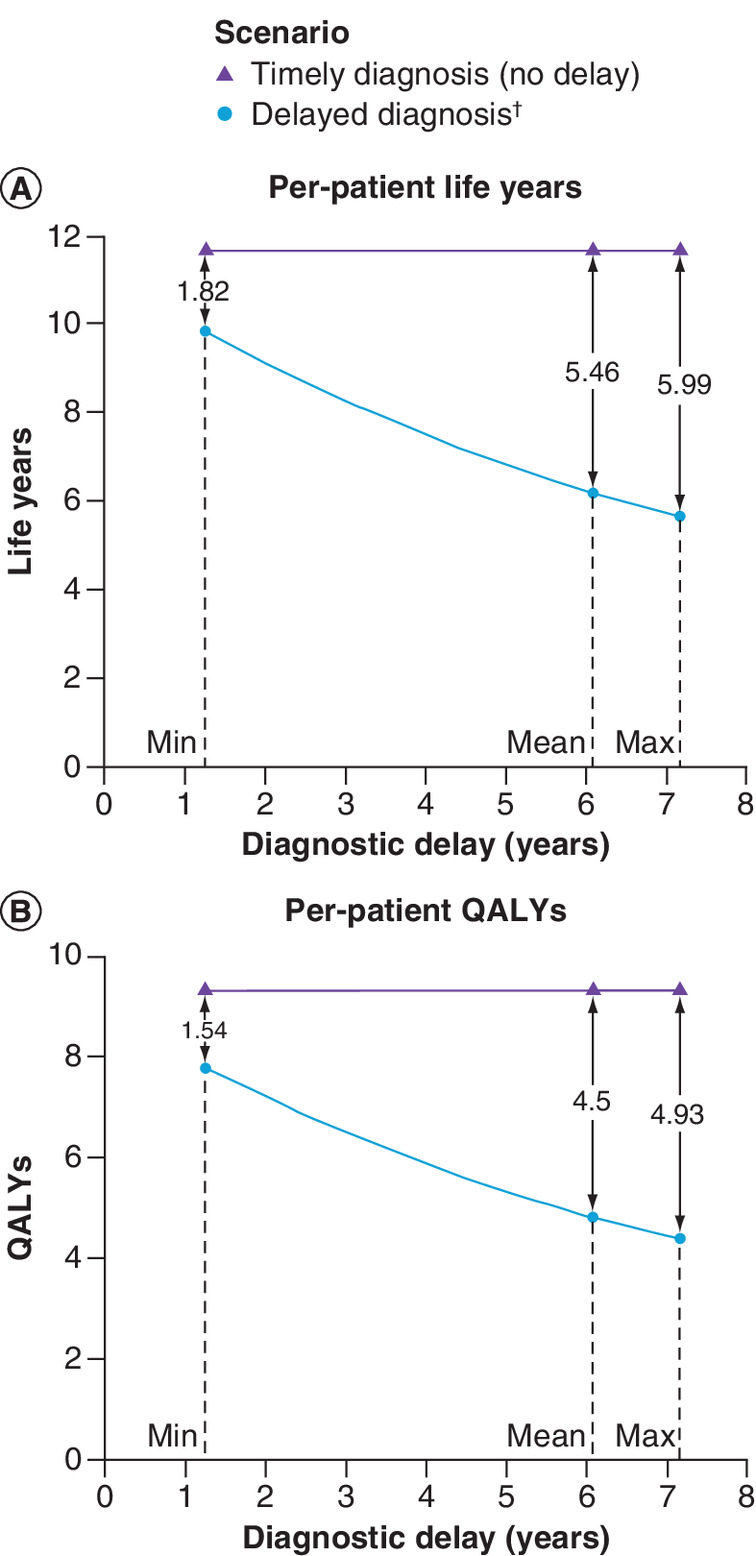
Figure 3. Per-patient health outcomes: ATTRwt-CM.
(A) ▲ markers denote life year predictions under the scenario of timely diagnosis and treatment; ● markers denote life year predictions for scenarios considering the mean and range diagnostic delays. Gains associated with timely diagnosis and treatment are shown as the difference between the respective points. (B) Per A for QALYs.
†Diagnostic delays correspond to those presented in Table 1.
QALY: Quality-adjusted life year.
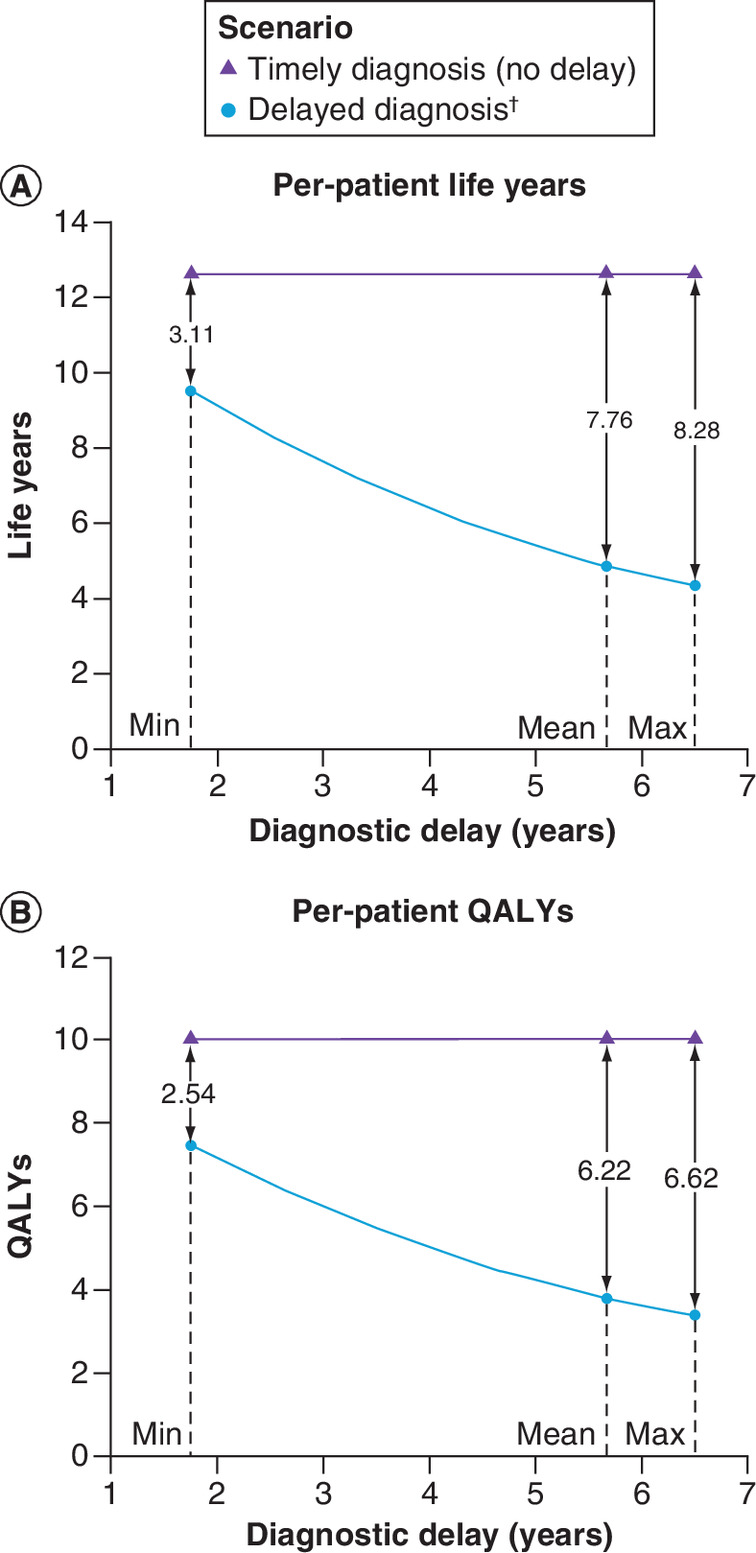
Figure 4. Per-patient health outcomes: ATTRv-CM.
(A) ▲ markers denote life year predictions under the scenario of timely diagnosis and treatment; ● markers denote life year predictions for scenarios considering the mean and range diagnostic delays. Gains associated with timely diagnosis and treatment are shown as the difference between the respective points. (B) Per A for QALYs.
†Diagnostic delays correspond to those presented in Table 1.
QALY: Quality-adjusted life year.
Sensitivity & scenario analyses
Figure 5 summarizes the impact of sensitivity and scenario analysis variations on incremental health outcomes. For both ATTR-CM types, timely diagnosis and treatment were predicted to improve health outcomes across all sensitivity and scenario analyses conducted.
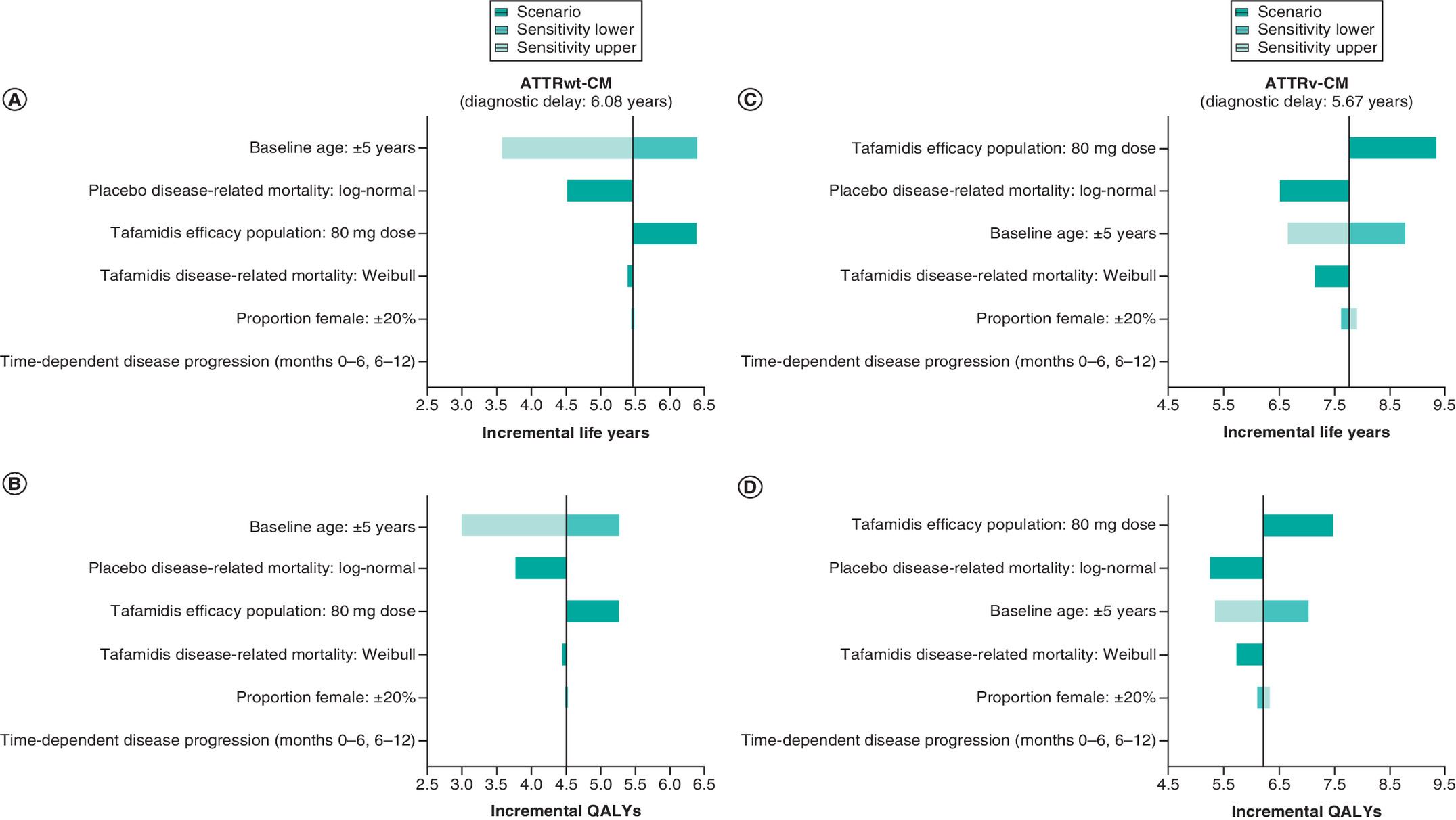
Figure 5. Sensitivity and scenario analyses: tornado plot of incremental health outcomes.
Incremental life years (A) and QALYs (B) predicted for ATTRwt-CM patients in the primary analysis are denoted by the vertical lines with the impact of variations made for sensitivity and scenario analyses depicted by the horizontal bars. Sensitivity and scenario analyses are ranked in order of impact. (C & D): As above for ATTRv-CM.
NYHA: New York Heart Association; QALY: Quality-adjusted life year.
Model predictions were sensitive to baseline age variations (±5 years), with greater health benefits predicted for younger patients and less benefit for older patients. Driven by improved survival outcomes, predicted health benefits were greater when tafamidis efficacy profiles were based on the 80 mg dosing arm alone; in the primary analysis, tafamidis efficacy profiles were based on the pooled (tafamidis meglumine 20 and 80 mg dosing) tafamidis arms. Variations to the sex distribution of the cohort (proportion female ±20%) had limited impact for either ATTR-CM type; however, the model results indicate greater benefit for females, with the proportionally greater differences predicted for ATTRv-CM patients due in part to the fact that this cohort contained a greater proportion of females than the ATTRwt-CM cohort (Table 1).
The use of alternative survival models (see S2.3 for detail) to characterize disease-related mortality also influenced results, providing more conservative estimates of the expected health gains for timely diagnosis and treatment than the primary analyses. Modeling only NYHA class transitions observed in each 6-month assessment interval in ATTR-ACT for the early assessment phases (months 0–6 and months 6–12) had a negligible impact on results.
Discussion
Diagnostic delay for ATTR-CM is widely acknowledged in the literature as a clinical challenge [6,11–13]. However, until the advent of disease-modifying treatment, its consequences for patients' life expectancy and HRQoL were limited. To our knowledge, the benefits of timely diagnosis and treatment in the era of disease-modifying treatment have not previously been quantified.
This study demonstrates that lengthy diagnostic delays represent a foregone opportunity to extend and improve life for patients with ATTR-CM through treatment with a disease-modifying therapy, tafamidis. Relative to delayed diagnosis, timely diagnosis and treatment for ATTRwt-CM patients were predicted to result in a mean life expectancy gain of 5.46 years and a mean QALY gain of 4.50. For ATTRv-CM patients, mean life expectancy and QALY gains were 7.76 and 6.22, respectively. Most of the benefit was accrued in the NYHA classes corresponding to less advanced heart failure (NYHA class I and II), with patients from the modeled ATTRwt-CM and ATTRv-CM cohorts predicted to spend markedly greater amounts of time in these classes under conditions of timely diagnosis and treatment than if they had experienced delay. These findings reflect the observation that, due to its mechanism of action and ATTR-CM's progressive nature, tafamidis is expected to provide greater benefit, both in overall extension of life and extension of life in lower heart failure classes, to patients who are treated earlier in the disease course [24,38].
Sensitivity analyses found that estimated benefits were greater for younger patients and females. The prediction that younger patients derive greater benefit further underscores the need for earlier diagnosis in order to maximize treatment benefit. Consistent with the findings of published post-hoc analyses of ATTR-ACT and its LTE [37], scenario analyses also found that estimated health benefits were greater when tafamidis efficacy was based on the ATTR-ACT 80 mg dosing arm alone. These findings suggest that estimates based on the efficacy of the pooled (tafamidis meglumine 20 and 80 mg) tafamidis dosing arms may be conservative, and further support FDA and EMA recommendations for use of the tafamidis meglumine 80 and the 61 mg free acid bioequivalent doses, respectively [22,23].
Our study did not consider the economic implications of diagnostic delay. However, there is evidence that patients with delayed diagnosis have high healthcare resource utilization during their diagnostic journey, typically making numerous healthcare visits, including inpatient hospitalizations, and undergoing a range of investigations for other conditions [12,26]. This cycling through different investigations and services would be largely avoided with prompt diagnosis. An economic evaluation by Pilgaard et al. of diagnostic and lifetime hospital costs of patients with ATTRwt-CM in the Danish healthcare setting concluded that healthcare resource use costs could be reduced through earlier diagnosis [39].
Our study has notable strengths. Treatment efficacy, quantified in terms of survival and NYHA class status, and HRQoL were informed by analyzing individual patient-level data from the pivotal trial (ATTR-ACT) and its LTE, which followed tafamidis-managed patients to a median treatment duration of 51.9 months [40]. This can be considered the best available evidence, given that ATTR-ACT was a Phase III randomized controlled trial providing direct evidence on the efficacy of tafamidis versus placebo. Modeled diagnostic delay and baseline demographic characteristic profiles were derived from a comprehensive literature review that captured reports of diagnostic delay from real-world clinical practice [13]. Furthermore, unlike studies evaluating the benefits of screening interventions, the findings of our study are not subject to lead time bias. Given that the evidence source informing outcome estimates in both the timely and delayed diagnosis and treatment scenarios is a single randomized controlled trial, and that survival predictions are made from a common point of disease development (as detailed in the baseline characteristics in Table 1), the confounding effects of the lead time associated with earlier diagnosis are not incurred.
Some limitations of the study should be noted. First, no evidence was available in the literature on diagnostic delay to inform NYHA class distributions at symptom onset, so the distributions observed at randomization in ATTR-ACT were assumed to be in place in the analyses. While benefits were seen across patients with baseline NYHA class I–III in ATTR-ACT, the current analyses considered only those patients with baseline NYHA class I or II. As such, the modeled outcomes can be considered representative for patients with early-stage disease. Second, it was necessary to extrapolate survival outcomes beyond the observed data, as many patients enrolled in ATTR-ACT were still alive at the time of data cut-off. Nevertheless, as survival extrapolations were based on ATTR-ACT and its LTE, the model predictions can be considered to be informed by the best available evidence.
Conclusion
Facilitating timely diagnosis is a key element in the drive to improve the management of patients with ATTR-CM, and this study adds further evidence to support the urgency of reducing diagnostic delay. Timely diagnosis and treatment with tafamidis are predicted to significantly improve survival and health-related quality of life outcomes for ATTRwt-CM and ATTRv-CM patients compared with the historical scenario of delayed diagnosis and lack of a disease-modifying treatment.
Future perspective
The availability of a disease-modifying treatment, together with recent consensus recommendations on non-invasive diagnostic techniques and suggested diagnostic ‘red flags’, are leading to increased awareness of ATTR-CM among clinicians. This trend is expected to continue, with the result that patients will, on average, be diagnosed earlier in the disease process, increasing the benefit derived from treatment.
•
Transthyretin amyloid cardiomyopathy (ATTR-CM) is a progressive and ultimately fatal disease caused by the accumulation of amyloid fibrils in the heart muscle.
•
Diagnostic delay is common, with patients often not correctly diagnosed for several years after symptom onset.
•
Since the recent introduction of the first disease-modifying treatment, tafamidis, diagnostic delay represents a missed opportunity for intervention to extend and improve life for patients with ATTR-CM.
•
This study evaluated the expected health benefits of timely diagnosis and treatment of ATTR-CM with tafamidis in the US setting, using a disease simulation model.
•
Analyses were underpinned by the findings of a targeted literature review and individual patient-level data from the pivotal Phase III trial, Tafamidis in Transthyretin cardiomyopathy Clinical Trial (ATTR-ACT).
•
Timely diagnosis and treatment were predicted to substantially improve survival and health-related quality of life outcomes for ATTRwt-CM and ATTRv-CM patients, compared with the historical scenario of delayed diagnosis and lack of a disease-modifying treatment.
•
Estimated benefits were greater for younger patients, underscoring the need for treatment earlier in the disease course in order to maximize benefit.
•
Facilitating timely diagnosis is a key element in the drive to improve the management of patients with ATTR-CM.
Financial & competing interests disclosure
This work was supported by Pfizer. MH Rozenbaum, S Large, R Bhambri and M Stewart are employees of Pfizer and own stock and/or stock options. R Young and A van Doornewaard are employees of Health Economics and Outcomes Research Ltd and received fees from Pfizer in relation to this study and development of the manuscript. J Nativi-Nicolau's institution received funding for clinical trials for Pfizer, Akcea and Eidos and Educational Grants from Pfizer. N Dasgupta has been a consultant for Pfizer, Ionis, Akcea and Alynlam. J Nativi-Nicolau has been a consultant for Pfizer, Eidos, Akcea and Alnylam. A Masri's institution received research grants from Pfizer, Akcea and Ultromics. A Masri has been a consultant for Eidos, Ionis and Cytokinetics. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Medical writing support was provided by Jo Whelan of Health Economics and Outcomes Research Ltd, funded by Pfizer.
Open access
This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
Supplementary Material
File (suppl_file.doc)
- Download
- 387.00 KB
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
1.
Ruberg FL, Grogan M, Hanna M, Kelly JW, Maurer MS. Transthyretin amyloid cardiomyopathy: JACC state-of-the-art review. JACC 73(22), 2872–2891 (2019).
•• Expert review of transthyretin amyloid cardiomyopathy (ATTR-CM) and its diagnosis.
2.
Maurer MS, Bokhari S, Damy T et al. Expert consensus recommendations for the suspicion and diagnosis of transthyretin cardiac amyloidosis. Circ. Heart Fail. 12(9), e006075 (2019).
•• Expert consensus recommendations on the diagnosis of ATTR-CM, including the use of the latest noninvasive techniques.
3.
Gillmore JD, Damy T, Fontana M et al. A new staging system for cardiac transthyretin amyloidosis. Eur. Heart J. 39(30), 2799–2806 (2017).
4.
Maurer MS, Hanna M, Grogan M et al. Genotype and phenotype of transthyretin cardiac amyloidosis: THAOS (Transthyretin Amyloid Outcome Survey). JACC 68(2), 161–172 (2016).
5.
Kroi F, Fischer N, Gezin A, Hashim M, Rozenbaum MH. Estimating the gender distribution of patients with wild-type transthyretin amyloid cardiomyopathy: a systematic review and meta-analysis. Cardiol. Ther. 10, 41–55 (2021).
6.
Witteles RM, Bokhari S, Damy T et al. Screening for transthyretin amyloid cardiomyopathy in everyday practice. JACC Heart Fail. 7(8), 709–716 (2019).
•• Guide to early diagnosis of ATTR-CM in clinical practice, including ‘red flags’ that should increase suspicion.
7.
Jacobson DR, Alexander AA, Tagoe C, Buxbaum JN. Prevalence of the amyloidogenic transthyretin (TTR) V122I allele in 14–333 African–Americans. Amyloid 22(3), 171–174 (2015).
8.
Quarta CC, Buxbaum JN, Shah AM et al. The amyloidogenic V122I transthyretin variant in elderly Black Americans. N. Engl. J. Med. 372(1), 21–29 (2014).
9.
Brunjes DL, Castano A, Clemons A, Rubin J, Maurer MS. Transthyretin cardiac amyloidosis in older Americans. J. Card. Fail. 22(12), 996–1003 (2016).
10.
Alpert CM, Smith MA, Hummel SL, Hummel EK. Symptom burden in heart failure: assessment, impact on outcomes, and management. Heart Fail. Rev. 22(1), 25–39 (2017).
11.
Rapezzi C, Lorenzini M, Longhi S et al. Cardiac amyloidosis: the great pretender. Heart Fail. Rev. 20(2), 117–124 (2015).
12.
Lane T, Fontana M, Martinez-Naharro A et al. Natural history, quality of life, and outcome in cardiac transthyretin amyloidosis. Circulation 140(1), 16–26 (2019).
• Natural history and outcomes of ATTR-CM in a large national cohort.
13.
Rozenbaum MH, Large Samuel, Bhambri Rahul et al. Impact of delayed diagnosis and misdiagnosis for patients with transthyretin amyloid cardiomyopathy (ATTR-CM): a targeted literature review. Cardiol. Ther. 10, 141–159 (2021).
•• Review of the literature on diagnostic delay in ATTR-CM; the findings form the basis of the delay durations modeled in the current study.
14.
Falk RH. Tafamidis for transthyretin amyloid cardiomyopathy: the solution or just the beginning of the end? Eur. Heart J. 40(12), 1009–1012 (2019).
15.
Gillmore JD, Maurer MS, Falk RH et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation 133(24), 2404–2412 (2016).
16.
Maurer MS. Noninvasive identification of ATTRwt cardiac amyloid: the re-emergence of nuclear cardiology. Am. J. Med. 128(12), 1275–1280 (2015).
17.
Koike H, Katsuno M. Transthyretin amyloidosis: update on the clinical spectrum, pathogenesis, and disease-modifying therapies. Neurol. Ther. 9(2), 317–333 (2020).
18.
ASNC Practice Points: 99mTechnetium-Pyrophosphate Imaging for Transthyretin Cardiac Amyloidosis (2018). www.asnc.org/content.asp?contentid=290
19.
Dorbala S, Ando Y, Bokhari S et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis: part 2 of 2-Diagnostic criteria and appropriate utilization. J. Nucl. Cardiol. 27(2), 659–673 (2020).
20.
Nativi-Nicolau J, Siu A, Dispenzieri A et al. Temporal trends of wild-type Attr amyloidosis in the Transthyretin Amyloidosis Outcomes Survey. J. Card. Fail. 26(10), S82 (2020).
21.
Lousada I, Maurer M, Warner M, Guthrie S, Hsu K, Grogan M. Amyloidosis research consortium cardiac amyloidosis survey: results from patients with AL and ATTR amyloidosis and their caregivers. Presented at: XVIth International Symposium on Amyloidosis; 2018 March 26–29 Kumamoto, Japan (2018).
22.
European Medicines Agency. Assessment report: vyndaqel (2019). www.ema.europa.eu/en/documents/variation-report/vyndaqel-h-c-2294-x-0049-g-epar-assessment-report_en.pdf
23.
US Food and Drug Administration. FDA approves new treatments for heart disease caused by a serious rare disease, transthyretin mediated amyloidosis (2019). www.fda.gov/news-events/press-announcements/fda-approves-new-treatments-heart-disease-caused-serious-rare-disease-transthyretin-mediated#:~:text=On%20May%203%2C%20the%20U.S.,approved%20treatments%20for%20ATTR%2DCM
24.
Maurer MS, Schwartz JH, Gundapaneni B et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N. Engl. J. Med. 379(11), 1007–1016 (2018).
25.
Bishop E, Brown EE, Fajardo J, Barouch LA, Judge DP, Halushka MK. Seven factors predict a delayed diagnosis of cardiac amyloidosis. Amyloid 25(3), 174–179 (2018).
26.
Ladefoged B, Dybro A, Povlsen JA, Vase H, Clemmensen TS, Poulsen SH. Diagnostic delay in wild type transthyretin cardiac amyloidosis – a clinical challenge. Int. J. Cardiol. 304, 138–143 (2020).
27.
TA267: ivabradine for treating chronic heart failure. National Institute for Health and Care Excellence (2012). www.nice.org.uk/Guidance/TA267
28.
TA388: sacubitril valsartan for treating symptomatic chronic heart failure with reduced ejection fraction. National Institute for Health and Care Excellence (2016). www.nice.org.uk/guidance/ta388
29.
Classes of Heart Failure (2017). American Heart Association. www.heart.org/en/health-topics/heart-failure/what-is-heart-failure/classes-of-heart-failure
30.
Chen J, Normand S-LT, Wang Y, Krumholz HM. National and regional trends in heart failure hospitalization and mortality rates for Medicare beneficiaries, 1998–2008. JAMA 306(15), 1669–1678 (2011).
31.
Dunlay SM, Redfield MM, Weston SA et al. Hospitalizations after heart failure diagnosis: a community perspective. JACC 54(18), 1695–1702 (2009).
32.
Ponikowski P, Voors AA, Anker SD et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. J. Heart Fail. 18(8), 891–975 (2016).
33.
Pfizer. ATTR-ACT, data on file (2019).
34.
Pfizer. ATTR-ACT extension study (Preliminary August 2019 database lock), data on file (2019).
35.
Arias E, Xu J. United States life tables: 2017. Centers for Disease Control and Prevention (2019).
36.
Shaw JW, Johnson JA, Coons SJJMC. US valuation of the EQ-5D health states: development and testing of the D1 valuation model. Med. Care 43(3), 203–220 (2005).
37.
Damy T, Garcia-Pavia P, Hanna M et al. Efficacy and safety of tafamidis doses in the Tafamidis in Transthyretin Cardiomyopathy Clinical Trial (ATTR-ACT) and long-term extension study. Eur. J. Heart Fail. 23(2), 277–285 (2020).
38.
Keohane D, Schwartz J, Gundapaneni B, Stewart M, Amass L. Tafamidis delays disease progression in patients with early stage transthyretin familial amyloid polyneuropathy: additional supportive analyses from the pivotal trial. Amyloid 24(1), 30–36 (2017).
39.
Pilgaard T, Pedersen MH, Poulsen SH. Diagnostic and lifetime hospital costs of patients suffering from wild-type transthyretin amyloid cardiomyopathy in Denmark. J. Med. Econ. 23(10), 1084–1091 (2020).
40.
Damy T, Elliott P, Gundapaneni B et al. Long-term survival with tafamidis in patients with transthyretin amyloid cardiomyopathy. Eur. Heart J. 41(Suppl. 2), ehaa946.2142 (2020).
Information & Authors
Information
Published In
Pages: 927 - 938
PubMed: 34142865
Copyright
© 2021 Pfizer Inc. This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 Unported License
History
Received: 18 March 2021
Accepted: 7 May 2021
Published online: 18 June 2021
Keywords:
Topics
Authors
Funding Information
Metrics & Citations
Metrics
Article Usage
Article usage data only available from February 2023. Historical article usage data, showing the number of article downloads, is available upon request.
Citations
How to Cite
Estimating the health benefits of timely diagnosis and treatment of transthyretin amyloid cardiomyopathy. (2021) Journal of Comparative Effectiveness Research. DOI: 10.2217/cer-2021-0071
Export citation
Select the citation format you wish to export for this article or chapter.
Citing Literature
- Carol Yen, John W. Epling, Michelle Rockwell, Monifa Vaughn-Cooke, Toward Safer Diagnoses: A SEIPS-Based Narrative Review of Diagnostic Errors, Diagnostics, 10.3390/diagnostics16020347, 16, 2, (347), (2026).
- Allen Chen, Re-evaluating the access imperative in healthcare in the United States, Journal of Public Health Policy, 10.1057/s41271-025-00612-7, (2026).
- Bruria Hirsh-Raccah, Treatment of Cardiac Amyloidosis—Disease Modifying Therapies in ATTR: Pharmacological Point of View, Cardiac Amyloidosis, 10.1007/978-3-031-88342-2_16, (261-283), (2025).
- Thibaud Damy, Rong Wang, Mathew S. Maurer, Julian D. Gillmore, Marianna Fontana, Long-Term Efficacy of Tafamidis in Patients with Transthyretin Amyloid Cardiomyopathy by National Amyloidosis Centre stage, European Journal of Heart Failure, 10.1002/ejhf.3696, 27, 12, (2998-3009), (2025).
- Laura De Michieli, Alessandro Lupi, Giulio Sinigiani, Angela Tietto, Alessandro Salvalaggio, Antonio Branca, Stefano Da Pozzo, Stefania Rizzo, Diego Cecchin, Martina Perazzolo Marra, Tamara Berno, Domenico Corrado, Chiara Briani, Alberto Cipriani, Pharmacological Management of Transthyretin Amyloid Cardiomyopathy: Where We Are and Where We Are Going, Journal of Clinical Medicine, 10.3390/jcm14103481, 14, 10, (3481), (2025).
- Allen M. Chen, Why Access Matters in Value-Based Healthcare: A Systematic Review, Journal for Healthcare Quality, 10.1097/JHQ.0000000000000471, 47, 2, (2025).
- Ahmad Masri, Yong Chen, A. Carmine Colavecchia, Darrin Benjumea, Aaron Crowley, Priti Jhingran, Matthew Kent, Jenifer Wogen, Cindi Pankratova, Jose Maria Jimenez Alvir, Rahul Bhambri, Coexisting Calcific Aortic Stenosis and Transthyretin Cardiac Amyloidosis: Real‐World Evaluation of Clinical Characteristics and Outcomes, Journal of the American Heart Association, 10.1161/JAHA.123.033251, 14, 2, (2025).
- Sergi Yun, Giovanni Palladini, Lisa J. Anderson, Eve Cariou, Ronnie Wang, Franca S. Angeli, Ben Ebede, Pablo Garcia-Pavia, International prevalence of transthyretin amyloid cardiomyopathy in high-risk patients with heart failure and preserved or mildly reduced ejection fraction, Amyloid, 10.1080/13506129.2024.2398446, 31, 4, (291-301), (2024).
- Sandra Michaela Ihne-Schubert, Maria Leberzammer, Marcel Weidgans, Stefan Frantz, Hermann Einsele, Stefan Knop, Torben Schubert, Tanja Bratan, Stefan Störk, Silke Neuderth, Single German centre experience with patient journey and care-relevant needs in amyloidosis: The German AMY-NEEDS research and care program, PLOS ONE, 10.1371/journal.pone.0297182, 19, 5, (e0297182), (2024).
- Anson T.C. Lau, Robert J. DiDomenico, Kibum Kim, Cost-effectiveness of systematic screening and treatment of transthyretin amyloid cardiomyopathy (ATTR-CM) in patients with heart failure with preserved ejection fraction (HFpEF) in United States, International Journal of Cardiology, 10.1016/j.ijcard.2023.131598, 398, (131598), (2024).