A criterion-based approach to systematic and transparent comparative effectiveness: a case study in psoriatic arthritis
Publication: Journal of Comparative Effectiveness Research
Abstract
Aim: Indirect treatment comparisons are used when no direct comparison is available. Comparison networks should satisfy the transitivity assumption, that is, equal likelihood of treatment assignment for a given patient based on comparability of studies. Materials & methods: Seven criteria were evaluated across 18 randomized controlled trials in psoriatic arthritis: inclusion/exclusion criteria, clinical trial design and follow-up, patient-level baseline characteristics, disease severity, prior therapies, concomitant and extended-trial treatment and placebo response differences. Results: Across studies, placebo was a common comparator, and key efficacy end points were reported. Collectively, several potential sources of insufficient transitivity were identified, most often related to trial design and population differences. Conclusion: Potential challenges in satisfying transitivity occur frequently and should be evaluated thoroughly.
Randomized controlled trials (RCTs) often compare new drugs with placebo [1,2]. In the absence of head-to-head studies comparing two active agents, indirect treatment comparisons (ITCs) with a common drug comparator can be used to estimate each drug's relative clinical effectiveness. For example, using a common treatment arm B, results from a clinical trial directly comparing drugs B and A can be indirectly compared with another, separate clinical trial directly comparing drugs B and C [2–4]. As shown in Supplementary Figure 1, with network meta-analyses (NMAs), multiple ITCs can be joined, either with or without direct comparisons (trials), as long as there is a common treatment comparator.
ITC is a valid and robust statistical approach when the transitivity assumption is met [1,4,5]. Within a treatment network containing either direct or both direct and indirect comparisons, the transitivity assumption states equal likelihood of receiving any of the treatments for each patient [3]. Assuming transitivity requires satisfaction of a complex standard that joins several points of consideration to compare study similarities (comparability) across the treatment network [1,4,5]. Although various discussions in the literature offer general advice on assessing studies for both conceptual and epidemiological differences, the authors did not find a single, centralized source in the literature to guide the evaluation of transitivity. For example, the findings from an NMA can be more precise when there is sufficient between-study similarity in clinical trial designs and balance of patient baseline characteristics that have potential treatment-modifying effects (i.e., effect modifiers). If these different study parameters and patient characteristics are effect modifiers, then combined study results can be biased, either overestimating or underestimating the significance of the relative treatment effect [2,6–8]. In these cases, guidelines from the literature suggest that ITC should not be performed, and transitivity can be considered unsatisfied [1,3,9]. Furthermore, if baseline study characteristics that are not effect modifiers are distributed too unevenly across studies (which can be evaluated with χ2 tests or t-tests, for example), then it can be challenging to infer comparative treatment effect estimates to broader patient populations [10,11]. These interpretive challenges do not necessarily invalidate the ITC; however, understanding the potential influence of the baseline differences across studies is clinically relevant [11]. When treating potentially different patient populations, this can be a strong theme during evaluation of transitivity.
Consistency, which represents the degree of difference between direct and indirect comparisons, can be used to help assess transitivity [12]. To satisfy consistency within the same network, estimates from direct comparison paths would approximate estimates from an indirect path. However, consistency can only be evaluated when there are both indirect and direct paths for the same pair of drugs being compared [3,12]. Furthermore, consistency alone only considers end results, not study and patient characteristics; thus, measuring consistency is not enough evidence to confirm transitivity. While statistical tests can help to gather information that demonstrate potential transitivity violations and challenges (e.g., significant imbalances in effect modifiers and clinical variables associated with prognosis), performing a comprehensive transitivity evaluation involves collecting multiple types of evidence. Thus, even if an ITC is technically feasible, the validity of ITCs should be rigorously evaluated during the review of the studies and before performing calculations.
In the last 15 years, the number of systematic reviews and NMAs has grown exponentially, including ITCs within clinical rheumatology [1,13,14]. With limited practical guidance to assess transitivity, networks are becoming increasingly large and complex, potentially including increased sources of heterogeneity across trials, which can be problematic. In the case of psoriatic arthritis (PsA), for example, there is a clear opportunity to develop networks of trials evaluating many treatments, including disease-modifying antirheumatic drugs (DMARDs) and biologic therapies consisting of TNF-α blockers and IL-12/23, IL-17 and IL-23 inhibitors (Supplementary Figure 1). It is less clear whether such comparisons would sufficiently address the transitivity requirement. We developed criteria to assess transitivity and applied these criteria to PsA trials, using the new treatment apremilast as an example case for an NMA, to better define the opportunities and challenges in conducting NMAs appropriately. By evaluating PsA studies for transitivity, we aimed to further refine existing discussion surrounding transitivity into practical and specific guidance to inform transitivity requirements for NMAs.
Many treatment options and variabilities exist in PsA drug sequencing based on patient characteristics and preference, disease manifestation and drug response. Pathways traditionally begin with systemic therapy (e.g., nonsteroidal anti-inflammatory drugs or DMARDs, such as methotrexate [MTX]), then vary according to response and tolerability. Biologic therapies for PsA, including but not limited to etanercept, infliximab, adalimumab or golimumab, target TNF and are administered by injections or infusions. The IL-12/23 and IL-17 biologic inhibitors act on different parts of the inflammatory cascade. Apremilast is an oral phosphodiesterase 4 (PDE4) inhibitor that regulates the inflammatory cascade [15]. In four randomized, placebo-controlled studies, apremilast treatment improved clinical disease outcomes in patients with active PsA [15,16]. To date, apremilast has not been compared directly with other monotherapies in head-to-head studies, including conventional DMARDs or biologics. At the time this analysis was performed, peer-reviewed published NMAs for PsA treatments did not include apremilast or they did not evaluate transitivity thoroughly [6,17–20]. Because head-to-head comparative evidence is lacking, a valid ITC of apremilast and its active comparators was chosen as a case study to evaluate another potential treatment option for PsA [21].
Materials & methods
Overview
To assess potential challenges in satisfying transitivity within a network of apremilast and other PsA therapies, several literature searches were performed, as summarized in Figure 1. Three distinct, targeted literature searches were conducted to source transitivity discussion, definitions, examples and stated criteria in peer-reviewed literature; any discussion of transitivity (including effect modification) in PsA studies and the broader rheumatology literature; and any assessment of transitivity in current NMAs comparing PsA clinical trials. From these targeted searches, any existing methods for evaluating transitivity, including clinical and patient factors contributing to insufficient transitivity, were organized into five general guidelines of transitivity. Next, a systematic literature search, following the Cochrane guidelines for systematic review, was performed to identify PsA studies to be included in the proposed NMA. Clinical and patient factors extracted from the selected studies were applied to the sourced transitivity descriptions to create more specific, centralized transitivity criteria. From these formalized transitivity criteria, the appropriateness of including the selected PsA studies in the NMA was reported.
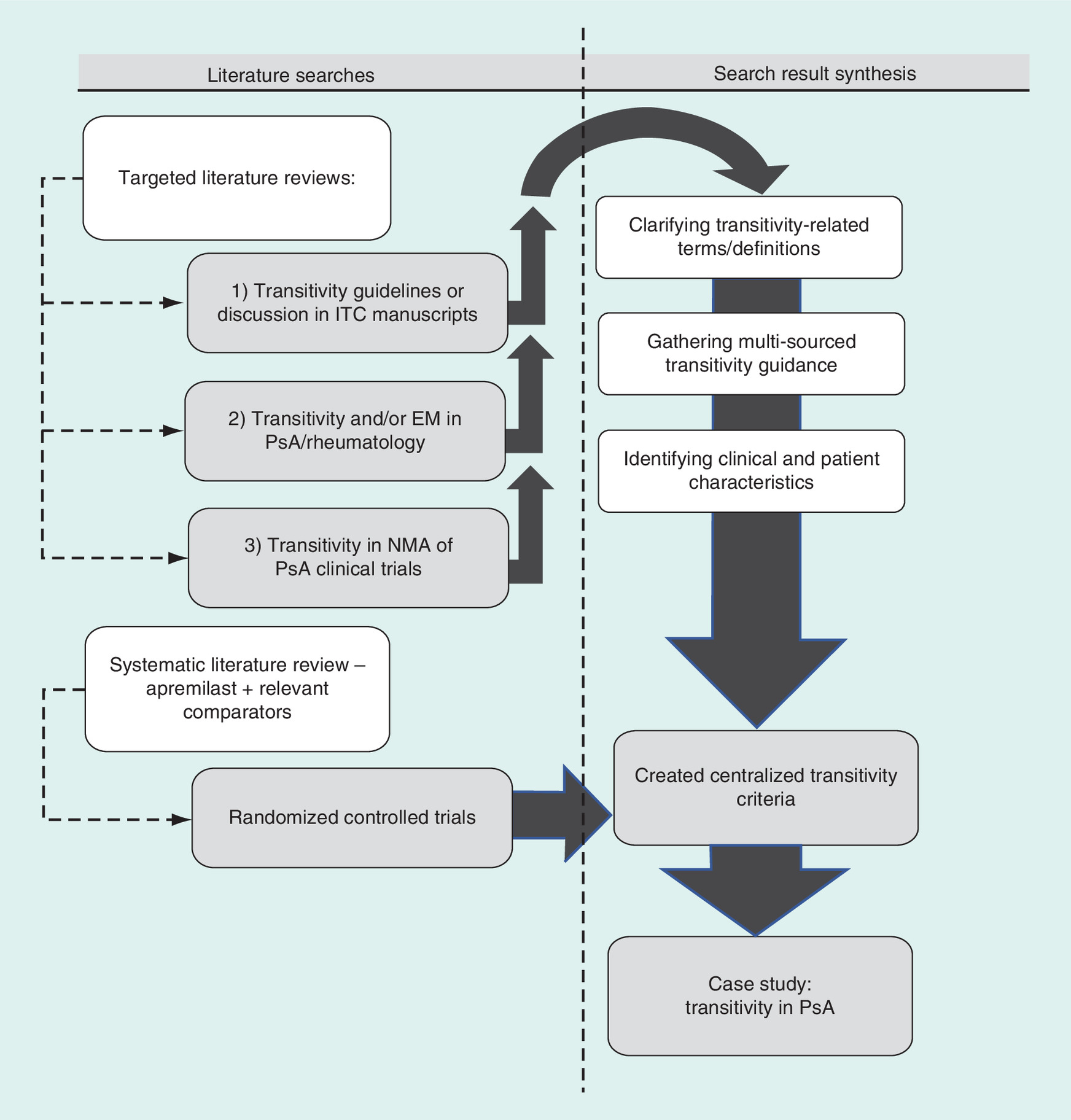
Figure 1. Overview of literature searches and evidence generation applied to newly centralized transitivity criteria.
EM: Effect modification; ITC: Indirect treatment comparison; NMA: Network meta-analysis; PsA: Psoriatic arthritis.
Search strategy & data extraction
The systematic literature search identified relevant studies in the databases Medline via PubMed, EMBASE, the Cochrane Central Register of Controlled Trials and clinicaltrials.gov. Eligible studies were RCTs of current PsA treatments published up to 1 March 2015. Treatments included in the search were MTX; the biologics adalimumab, certolizumab pegol, etanercept, golimumab, infliximab, secukinumab and ustekinumab; and the PDE4 inhibitor apremilast.
The Cochrane guidelines were followed during the selection of full-text articles [22]. At least two reviewers selected the articles from a title and abstract review, and a third reviewer was enlisted for any disagreements. English-language studies needed to report at least one of the key efficacy or safety end points and include separate reporting of patients who were at least 18 years of age. Exclusion criteria eliminated nonrandomized clinical studies, single-arm studies, studies without a control arm (placebo) and studies without full-text publications. A PRISMA flow diagram for study selection was created (Supplementary Figure 2).
To evaluate the RCTs against the newly centralized transitivity criteria, the following study characteristics were extracted: inclusion criteria, exclusion criteria, drug comparator, participant age, sex, duration of PsA disease in years, mean tender joint count at study baseline, prior use of DMARDs or biologics, time of efficacy assessment/crossover design, concomitant DMARD or MTX use and placebo response rates based on the American College of Rheumatology response criteria (ACR20 and ACR50). Also, it was recorded if the study authors evaluated each of these variables for potential treatment effect modification or prognostic significance and what their methods were for addressing effect modification and prognostic factors.
Transitivity in the literature
From the targeted literature searches, it was found that the term transitivity was associated with similarity assumption or used interchangeably with comparability [4,23–27]. The similarity assumption was used to describe both finding a true (i.e., similar) treatment effect between two interventions across multiple studies as well as used to describe a network having similar characteristics between studies. However, for the similarity assumption and discussions of comparability, no formal definition or criteria to assess appropriateness of an ITC were found. Furthermore, applying concepts of similarity or comparability alone can lead to a narrower understanding that all study and patient characteristics must be mostly uniform to assume transitivity. In fact, valid ITCs can be performed as long as there is similar distribution of the mutual effect modifiers between studies [1,5,12,27]. From this literature, effect modification is defined as the presence of one or more variables that changes the effect estimate for treatment on a given scale of measure [28]. Specifically, the alteration in treatment effect is different within different subgroups of the same patient characteristic, which may be statistically demonstrated through a test for interaction. Statistical interaction is considered if the combined effect of two factors, such as PsA treatment and PsA disease subtype, is greater than would be expected by the sum or multiplication of their independent effects [29]. Across studies, similar distribution of effect modifiers can be constructed through population-adjustment methods, including propensity score weighting and simulated treatment comparison, if at least one individual, patient-level dataset is available for adjustment [28]. However, addressing effect modification is only one of the five different guiding principles found from different sources during the targeted literature searches that describe transitivity [1,5,30]. Ideas related to transitivity were found from different literary sources and organized below into the five general guidelines.
Current transitivity discussions found in the literature
Search results are organized into five guiding themes: between studies, there exists an anchor treatment, such as a placebo, that is similar in all studies being compared; the selection of studies for the network should not be solely motivated by an expected outcome, or results can be subject to bias; there are true comparative treatment effects between two interventions that can be measured across multiple studies, so that a network of multiple studies is justified; similar distribution of effect modifiers is necessary between studies, such as proportion of older versus younger patients, or poorer versus fitter baseline performance status; and any of the treatments could be appropriately compared in a randomized study based on similar disease indication.
Results
Evidence network
After title and abstract review, 82 publications were selected for full-text review. Some studies were associated with more than one publication, which resulted in the identification of 25 unique clinical trials (Supplementary Figure 2). Applying the inclusion and exclusion criteria and designating placebo as the comparator for a potential NMA, we were able to link 18 unique RCTs (Table 1) [15,16,31–46] for apremilast and seven active comparators (adalimumab, certolizumab pegol, etanercept, golimumab, infliximab, secukinumab or ustekinumab). Of note, two studies of MTX monotherapy were excluded because they lacked the required clinical response variables [47,48], and one apremilast abstract from 2014 (PALACE 4) was excluded for a similar lack of needed extraction variables [49]. During analysis, additional results from McInnes et al. for secukinumab and two follow-up studies for apremilast [15,16,31] were discovered and added. A graphic of the final network is provided in Supplementary Figure 1.
| Name | Study (year) | Treatment | Comparator | Ref. |
|---|---|---|---|---|
| PALACE 1 | Kavanaugh (2014) | Apremilast 20 mg OR 30 mg B.I.D. | Placebo | [33] |
| PALACE 2 | Cutolo (2016) | Apremilast 20 mg OR 30 mg B.I.D. | Placebo | [15] |
| PALACE 3 | Edwards (2016) | Apremilast 20 mg OR 30 mg B.I.D. | Placebo | [16] |
| PSA-001 | Schett (2012) | Apremilast 20 mg B.I.D. OR 40 mg QD | Placebo | [34] |
| ADEPT | Mease (2005) | Adalimumab 40 mg Q2W | Placebo | [39] |
| M02-570 | Genovese (2007) | Adalimumab 40 mg Q2W | Placebo | [37] |
| RAPID-PsA | Mease (2014) | Certolizumab pegol 200 mg Q2W OR Certolizumab pegol 400 mg Q4W | Placebo | [36] |
| Mease (2000) | Mease (2000) | Etanercept 25 mg twice weekly | Placebo | [40] |
| Mease (2004) | Mease (2004) | Etanercept 25 mg twice weekly | Placebo | [41] |
| GO-REVEAL | Kavanaugh (2009) | Golimumab 50 mg OR 100 mg Q4W | Placebo | [38] |
| IMPACT | Antoni (2005) | Infliximab 5 mg/kg weeks 0, 2, 6 and 14 | Placebo | [45] |
| IMPACT 2 | Antoni (2005) | Infliximab 5 mg/kg weeks 0, 2, 6 and 14 | Placebo | [42] |
| PSUMMIT 1 | McInnes (2013) | Ustekinumab 45 mg OR 90 mg at weeks 0 and 4, and every 12 weeks thereafter | Placebo | [35] |
| PSUMMIT 2 | Ritchlin (2014) | Ustekinumab 45 mg OR 90 mg at weeks 0 and 4, and every 12 weeks thereafter | Placebo | [32] |
| Gottlieb (2009) | Gottlieb (2009) | Ustekinumab 90 mg OR 63 mg QW | Placebo | [43] |
| Atteno (2010) | Atteno (2010) | Etanercept 25 mg twice weekly OR infliximab 5 mg/kg every 6–8 weeks | Adalimumab 40 mg Q2W | [46] |
| McInnes (2014) | McInnes (2014) | Secukinumab 10 mg/kg Q3W (two doses) | Placebo | [44] |
| FUTURE 2 | McInnes (2015) | Secukinumab 75 mg OR 150 mg OR 300 mg QW to week 4, then Q4W | Placebo | [31] |
B.I.D.: Twice per day; QD: Daily; QW: Weekly; Q2W: Every 2 weeks; Q3W: Every 3 weeks; Q4W: Every 4 weeks.
Development of the centralized transitivity criteria
Extraction of the patient and clinical trial characteristics of the selected PsA RCTs helped to identify specific variables that should be evaluated for transitivity across studies. This identification supported the development of the following transitivity criteria, with the aim of providing newly formalized, specific guidance. These centralized transitivity criteria were enlisted to determine if an ITC of the linked PsA RCTs could be appropriately performed. The seven transitivity criteria follow:
•
Inclusion/exclusion criteria: study participants should have similar trial eligibility, such as the same disease and indications for treatment, where they could be randomized to any of the trials in the network [1,4,50]. At the same time, exclusion criteria for comorbidities such as concurrent malignancies and infections should be comparable between studies [31,51,52].
•
Clinical trial design and follow-up: clinical trial designs should be comparable [4], with similar treatment durations [3], methods for identifying and measuring treatment outcomes [9] and length of follow-up periods [9]. For example, treatment crossover (switching) prior to the initial efficacy assessment introduces bias in measuring true effect depending how data are handled postswitching [53].
•
Baseline characteristics: patient factors such as age [3,54] and duration of disease [21] can modify relative treatment effects within PsA and thus should have similar distributions between all study arms to minimize biases in estimating effect size [1,4,9]. During randomization, patients may be stratified by potential prognostic factors that are directly associated with outcomes such as survival and disease response.
•
•
•
•
Placebo response: placebo groups may have higher than expected clinical responses, which can be associated with measuring lower relative treatment effects [57] and can convey the presence of confounding by unadjusted baseline risks or clinical trial differences [1,5,58]. These sources of bias can be inconsistent across different study protocols.
Data extraction & transitivity evaluation
Among the selected studies, sufficient pre-identified study variables were available to perform cross-study comparisons (Table 2) [15,16,31–46]. The apremilast trials (PALACE 1 [33], PALACE 2 [15] and PALACE 3 [16]) were considered the reference studies used to evaluate transitivity in comparison with the remaining studies in the network. Patients from these studies were permitted past exposure to biologics and DMARDS and could have concomitant use of MTX, sulfasalazine, leflunomide, low dose oral corticosteroid or nonsteroidal anti-inflammatory drugs during the studies. Further study data that supported the identification of transitivity violations are summarized in Table 2.
| Name | Study (year) | Age | Percentage of males | PsA duration, years | Baseline TJC | Cross-over | Percentage of prior biologic use | Percentage of prior DMARD | Percentage of concomitant DMARD use | Percentage of concomitant MTX use | Percentage of ACR-20 in placebo | Percentage of ACR-50 in placebo | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PALACE 1 | Kavanaugh (2014) | 49–51 | 45–52 | 7.2–8.1 | 22–23 | Week 16 | 22–24 | 96–100 | 63–66 | 52–57 | 19 | 6 | [33] |
| PALACE 2 | Cutolo (2016) | 51 | 42–47 | 6.8–7.8 | 18–22 | Week 16 | 14–17 | 97–100 | 70–71 | 58–70 | 19 | 5 | [15] |
| PALACE 3 | Edwards (2016) | 50 | 46–47 | 6.8–7.7 | 18–21 | Week 16 | 26–30 | 99–100 | 60–62 | 50–54 | 18 | 8 | [16] |
| PSA-001 | Schett (2012) | 50–51 | 47–62 | 7.3–8.4 | 21–23 | Week 12 | NR | NR | 43–45 | 43–45 | 12 | 3 | [34] |
| ADEPT | Mease (2005) | 49 | 55–56 | 9.2–9.8 | 24–26 | No | 0 | NR | 50–51 | 50–51 | 14 | 4 | [39] |
| M02-570 | Genovese (2007) | 48–50 | 51–57 | 7.2–7.5 | 25–29 | No | 0 | 100 | 65–67 | 47 | 16 | 2 | [37] |
| RAPID-PsA | Mease (2014) | 47–48 | 42–46 | 7.9–9.6 | 20–22 | Week 16 | 17–23 | 100 | 65–74 | 62–65 | 24 | 11 | [36] |
| Mease (2000) | Mease (2000) | 44–46 | 53–60 | 9.0–9.5 | 20–21 | No | NR | NR | 47 | 47 | 13 | 3 | [40] |
| Mease (2004) | Mease (2004) | 47–48 | 45–57 | 9.0–9.2 | NR | No | NR | NR | 42 | 41–42 | 16 | 4 | [41] |
| GO-REVEAL | Kavanaugh (2009) | 46–48 | 59–61 | 7.2–7.7 | 22–24 | Week 16 | 0 | 66–73 | NR | 47–49 | 9 | 9 | [38] |
| IMPACT | Antoni (2005) | 45–46 | 58 | 11.0–11.7 | 20–24 | Week 16 | NR | 100 | 64–79 | 46–65 | 10 | 0 | [45] |
| IMPACT 2 | Antoni (2005) | 47 | 51–71 | 7.5–8.4 | 25 | Week 16 | 0 | NR | 45–47 | 45–47 | 11 | 3 | [42] |
| PSUMMIT 1 | McInnes (2013) | 47–48 | 47–49 | 7.2–8.5 | 18–22 | Week 16 | NR | NR | NR | 47–50 | 23 | 9 | [35] |
| PSUMMIT 2 | Ritchlin (2014) | 48 | 52–57 | 3.4–4.9 | 23–27 | Week 12/16 | 55–60 | NR | NR | NR | 44 | 18 | [32] |
| Gottlieb (2009) | Gottlieb (2009) | 48–50 | 53–59 | 4.9–6.2 | 16–20 | Week 16 | 24–31 | 59–63 | 20–21 | 20–21 | 14 | 7 | [43] |
| Atteno (2010) | Atteno (2010) | 48–49 | 38–41 | NR | 12–13 | No | 0 | 100 | NR | 30–90 | NR | NR | [46] |
| McInnes (2014) | McInnes (2014) | 47–48 | 32–43 | 5.4–6.3 | 23–24 | No | 23–42 | NR | 43–47 | 43 | NR | NR | [44] |
| FUTURE 2 | McInnes (2015) | 47–50 | 49–55 | NR | 20–24 | Week 16 | 33–37 | NR | NR | 44–51 | 15 | 7 | [31] |
ACR: American College of Rheumatology proportional improvement (20%, 50%); DMARD: Disease-modifying antirheumatic drug; MTX: Methotrexate; NR: Not reported; PsA: Psoriatic arthritis; TJC: Tender joint count.
Data extraction revealed potential sources of between-study clinical and methodological heterogeneity, and most study authors did not report statistical evaluation of treatment effect modification within their studies. Across the network, transitivity violations related to all seven tenets were found, and major themes included clinical trial design (crossover, timing of efficacy assessment and follow-up) and population differences (baseline disease characteristics, prior and concomitant medications and placebo response rates). Table 3 summarizes the most evident transitivity violations (or none) for each included study [15,16,31–46].
| Name | Study (year) | Summary | Ref. |
|---|---|---|---|
| PALACE 1 | Kavanaugh 2014 | No deviation (reference for comparison) | [33] |
| PALACE 2 | Cutolo (2016) | No deviation (reference for comparison) | [15] |
| PALACE 3 | Edwards (2016) | No deviation (reference for comparison) | [16] |
| PSA-001 | Schett (2012) | Structural: 12-week study vs 16-week PALACE studies | [34] |
| ADEPT | Mease (2005) | Structural: no crossover design; population – prior medications | [39] |
| M02-570 | Genovese (2007) | Structural: no crossover design; population – prior medications | [37] |
| RAPID-PsA | Mease (2014) | No significant deviations | [36] |
| Mease (2000) | Mease (2000) | Structural: no crossover design | [40] |
| Mease (2004) | Mease (2004) | Structural: no crossover design; population – missing variables on prior medications | [41] |
| GO-REVEAL | Kavanaugh (2009) | Population: prior biologic use excluded | [38] |
| IMPACT | Antoni (2005) | Population: PsA duration | [45] |
| IMPACT 2 | Antoni (2005) | Population: prior biologic use excluded | [42] |
| PSUMMIT 1 | McInnes (2013) | Missing variables on prior and concomitant medications | [35] |
| PSUMMIT 2 | Ritchlin (2014) | Population: PsA duration shorter, prior biologic use with lower clinical responses | [32] |
| Gottlieb (2009) | Gottlieb (2009) | Lower concomitant DMARDs use | [43] |
| Atteno (2010) | Atteno (2010) | Structural: no crossover design; population – lower TJC, exclusions for biologics; study design: missing data on concomitant medications | [46] |
| McInnes (2014) | McInnes (2014) | Structural: no crossover design, other concomitant medications | [44] |
| FUTURE 2 | McInnes (2015) | Missing variables on PsA duration, prior and other concomitant medications | [31] |
DMARD: Disease-modifying antirheumatic drug; PsA: Psoriatic arthritis; TJC: Tender joint count.
Study differences in data reported that contributed to transitivity violations were especially notable when comparing apremilast with the biologic studies. For instance, the apremilast studies included treatment crossover (early escape), enabling nonresponders in the placebo groups to be re-randomized [15,16,33,34]. However, efficacy end points occurred prior to re-randomization, which limited a potential source of measurement bias. Of the biologic studies, six did not report a crossover design (Transitivity Criteria #2); however, two studies permitted early escape to investigational treatment after initial treatment response measurements. Of the eight biologic studies that did have patient crossover, different methods were used to account for missing variables, making them difficult to compare. Of note, it is possible that crossover or re-randomization was performed but not explicitly reported in the article. As shown in Table 2, duration of PsA disease at baseline ranged from 3 years to nearly 12 years (Transitivity Criteria #3), while the literature demonstrates that outcomes are less favorable with a longer PsA symptom duration or time to diagnosis [59–61]. Across studies, mean tender joint counts at baseline ranged from 12 to 29 (Transitivity Criteria #4). Almost all participants in three of the four apremilast studies (PALACE 1, 2 and 3 [15,16,33] but not PSA-001 [34]) had prior conventional DMARD use, while 43–71% in all four apremilast studies had concomitant DMARD use [15,16,33,34]. Six of the nonapremilast studies also distinguished between proportions of patients with prior (59–100%) and concomitant (20–90%) use, while it was unclear if the remaining studies distinguished between prior and concomitant use (Transitivity Criteria #5). Concomitant treatments were similarly heterogenous (e.g., MTX use 20–90%) across all studies (Transitivity Criteria #6), as was placebo response (9–44%), measured as ACR20 (Transitivity Criteria #7). Furthermore, many studies did not report prior or concomitant medication use. In addition, there was variability in the years the studies were published and the countries from which patients were recruited. While a specific measured variable representing access issues was not available, year of publication or country of study can convey different accessibilities based on the treatment's availability in a healthcare system at that time. These factors may have resulted in differences such as, but not limited to, disease severity, prior therapies received and concomitant medications, making it difficult to compare results across studies. In summary, different inclusion and exclusion criteria may have been used across studies.
Effect modification
Within most of the extracted RCTs, authors enlisted various methods to minimize baseline imbalances in patient, disease and treatment characteristics (Table 4) [15,16,31–46]. Although none of the study authors used the terms ‘prognostic factor’ or ‘effect modifier’ when reporting stratification, 15 of the 18 RCTs specified stratification during randomization. Participants were stratified according to baseline use of DMARDs [15,16,32–35,37–42], prior exposure to biologics [31,36,43] or no stratification factor was reported [44–46]. Of the studies not reporting stratification, all but one reported performing subgroup analyses to determine any difference in treatment efficacy by patient characteristic [44,46].
| Name | Study (year) | Stratification | Evidence reporting | Additional investigator observations | Ref. |
|---|---|---|---|---|---|
| PALACE 1 | Kavanaugh (2014) | Baseline DMARD use | Graphical and numeric reporting of higher treatment response in biologic naive Textual refutation of DMARD use | Regardless of biologic history, treatment statistically superior over placebo Patient characteristics were consistent across biologic history | [33] |
| PALACE 2 | Cutolo (2016) | Baseline DMARD use | Textual refutation of prior biologic and baseline DMARD use | Lower treatment efficacy is noted in nonconcomitant DMARD use but not statistically tested | [15] |
| PALACE 3 | Edwards (2016) | Baseline DMARD use | Statistical testing and refutation (supplement) of prior biologic and baseline DMARD use | Similar treatment efficacy regardless of other medications Highest but not statistically superior response in biologic naive | [16] |
| PSA-001 | Schett (2012) | Baseline MTX use | Numeric refutation of baseline MTX use Numeric conveyance of different treatment efficacy by PsA subtype | Small patient subgroups challenged statistical testing | [34] |
| ADEPT | Mease (2005) | Baseline MTX use | Statistical testing and refutation of baseline MTX use and extent of psoriasis BSA | Proportion of improvement similar regardless of MTX use through week 24 Concludes that interaction of treatment and MTX to be determined Exclusion criteria for any other DMARD | [39] |
| M02-570 | Genovese (2007) | Baseline DMARD use | Statistical testing and refutation of baseline DMARD use | Response in the treatment arm was reached for 15 of 29 men (52%) and 5 of 22 women (23%) | [37] |
| RAPID-PsA | Mease (2014) | Prior TNFi exposure and investigational site | Graphical and numeric refutation of prior TNFi exposure Numeric refutation of concomitant DMARD use | Somewhat dissimilar proportions of response by prior DMARD (supplement) | [36] |
| Mease (2000) | Mease (2000) | Baseline MTX use | Textual refutation of treatment interaction by baseline MTX use | Subgroup analyses showed no differences in treatment efficacy by MTX or corticosteroid use, nor by baseline PASI score (data not shown) | [40] |
| Mease (2004) | Mease (2004) | Baseline MTX use | Textual refutation of sex and baseline MTX use | No significant differences in response were observed from sensitivity analyses of subgroups | [41] |
| GO-REVEAL | Kavanaugh (2009) | Baseline MTX use | Statistical testing and refutation of baseline MTX use | − | [38] |
| IMPACT | Antoni (2005) | Not reported | Statistical testing and refutation of baseline DMARD or specific MTX use | − | [45] |
| IMPACT 2 | Antoni (2005) | Baseline MTX use and investigational site | Textual refutation of baseline DMARD or specific MTX use | Small patient group sizes may have made higher level responses to appear more frequently in non-MTX users | [42] |
| PSUMMIT 1 | McInnes (2013) | Baseline MTX use | Graphical and numeric refutation of baseline MTX use | Crossover at week 16, numeric reporting of efficacy starts at week 24 | [35] |
| PSUMMIT 2 | Ritchlin (2014) | Baseline MTX use | Graphical and numeric conveyance of higher placebo response in MTX users and anti-TNF naive statistical refutation of anti-TNF exposure | Crossover at week 16, numeric reporting of efficacy starts at week 24 | [32] |
| Gottlieb (2009) | Gottlieb (2009) | Prior anti-TNF exposure and investigational site | Textual reporting of adjustment during analyses for anti-TNF exposure | Patients with anti-TNF exposure were limited to about 20% of the study population | [43] |
| Atteno (2010) | Atteno (2010) | Not reported | Not reported | While stratification was not reported, exclusion for prior anti-TNFi exposure | [46] |
| McInnes (2014) | McInnes (2014) | Not reported | Graphical and numeric reporting of differences in treatment efficacy by TNFi exposure | Small patient subgroups challenged statistical testing Treatment effect appears greater in TNFi naive | [44] |
| FUTURE 2 | McInnes (2015) | Prior TNFi use | Statistical refutation of prior TNFi exposure and treatment interaction Textual refutation of MTX use | Magnitude of response higher in TNFi naive, but both exposure groups demonstrated significant treatment effects | [31] |
DMARD: Disease-modifying antirheumatic drug; MTX: Methotrexate; PASI: Psoriasis area and severity index; PsA: Psoriatic arthritis; TNFi: Tumor necrosis factor inhibitor.
During analyses, nine studies [15,16,33–35,37–40], as identified in Table 4, specified testing separate treatment effects by baseline MTX or DMARD use with Mantel–Haenszel stratification or analysis of variance regression, including the apremilast studies. However, posttrial, only two studies specifically addressed (and refuted) the significance of a statistical interaction term [34,44]. Of the remaining 16 studies that were included, there were varying levels of reporting rigor for subgroup analyses addressing more general treatment-modifying effects [15,16,31–33,35–43,45,46]. For at least one baseline variable, four studies provided statistical refutation of effect modification [16,37–39], one study [34] reported similar numeric proportions of responders between subgroups, five studies provided similar evaluation plus graphical evidence [32–36] and five studies [15,40,41,43,45] stated in the text that differences in treatment effects by subgroups were not evident. One study [46] did not report analyses for potential baseline imbalances. Some study authors, as noted in Table 4, cited smaller patient populations as challenging their ability to statistically test subgroups [34,42,44].
While most studies reported stratification for one patient characteristic, effect modification was not refuted for remaining variables. As previously noted, effect modification is detected on a specific scale (e.g., additive treatment effect); however, none of the studies specified the models used to estimate efficacy. Some studies [31,33,36,44], as described in Table 4, conveyed potentially higher treatment efficacy in patients unexposed to certain prior therapies. For example, Kavanaugh et al. [33] stratified participants by baseline use of DMARDs and graphically depicted potential effect modification for past exposure to biologic agents. The authors noted relatively lower clinical responses in biologic-experienced patients when compared with biologic-naive patients receiving apremilast or placebo [33]. Similarly, Mease et al. [36] stratified participants by prior TNF inhibitor exposure and reported somewhat dissimilar proportions of clinical response by concomitant DMARDs, which potentially demonstrated effect modification (Supplementary Material). Most study authors did not statistically evaluate treatment-effect modification within their studies, making it difficult to determine within-study, treatment-modifying biases. On the other hand, if an effect modifier was identified at the trial level, the distribution of this variable could be assessed across the network.
Separate from the systematic literature review, our targeted literature search for developing transitivity criteria found studies and published guidelines addressing effect modification within PsA and rheumatoid arthritis (RA) disease studies. Actual detection of effect modification was demonstrated in three RA studies, and effect modifiers included age, swollen joint count, prior DMARD use, concomitant DMARD use, concomitant MTX use and baseline risk (placebo effect) [21,26,29]. Additionally, Christensen et al. [21] potentially detected a modification of the treatment effect based on the disease duration but were unable to demonstrate its independent relative effects from prior DMARD use. Additional PsA, RA and NMA quality guidelines discuss and encourage testing for potential sources of effect modification [5] such as patient comorbidities [51,52] and gender (Consensus Working Party 2013); however, none of the extracted RCTs tested against these potential biases. Furthermore, two full publication NMAs that included apremilast cited variations in study populations and a lack of covariate adjustment in the meta-regressions as limitations; however, potential biases related to transitivity were not measured [18,20]. Within our network, the PALACE 4 abstract was excluded for lack of the reporting variables traditionally available for full publications, while both full publication NMAs included the PALACE 4 abstract in their analysis. Another NMA, available as an abstract, aimed to test treatment efficacy of several treatments, including apremilast, according to biologic-exposed and biologic-naive subgroups [19]. However, due to limited data, trends within the biologic-exposed subgroup could not be developed. Another abstract reporting similar NMA methodology also performed separate analyses by biologic status, although no other patient characteristics were identified [62].
Discussion
The systematic literature review for PsA RCTs for systemic treatments or monotherapies resulted in a potential network of 18 unique RCTs with placebo comparators. Key patient, disease, treatment and response variables were extracted and evaluated for potential biases related to uneven distribution of potential effect modifiers and other transitivity violations. Comprehensive transitivity criteria were developed from evaluating study differences between the 18 RCTs and a targeted literature review of existing guidelines and relevant disease publications.
Based on existing NMA quality guidelines, past transitivity descriptions and our own transitivity criteria, there would be numerous challenges concerning comparability before conducting an ITC among the extracted PsA RCTs. While most study methods included stratification on a select baseline covariate, other potential sources of transitivity violations, including the clinical trial structure and design (crossover, length of study), were still evident. Post-trial, most authors discussed at least one method of subgroup analyses to evaluate study imbalances; however, only five studies included statistical evidence. Although sensitivity analyses and/or adjusting for confounding while performing an NMA may help to balance study and patient characteristics, if excluding studies, the quantity of information lost to achieve sufficient transitivity could limit the interpretative results. This case study of PsA highlights the growing trend of publishing NMAs within clinical rheumatology, while guidance to address transitivity is still under development and not routinely applied [12,13].
With the lack of specific guidance to assess transitivity, existing NMAs for PsA and RA often assume transitivity to perform comparisons [1,4,5,9]. While guidelines in the literature addressing quality issues in NMA are increasing, such as the Grading of Recommendations Assessment, Development and Evaluation (GRADE) [30] and criteria by the Comparing Multiple Interventions Methods Group (Cochrane), our transitivity criteria provide specific and necessary detail to systematically check key study variables for between-study comparability when considering ITC.
Limitations
Transitivity considerations are limited by the ability to identify sources of heterogeneity based on individual trial reporting. Often there is potential for unobserved sources of bias or unreported study variables that limit valid comparisons between studies [1]. Particularly in the case of effect modifiers, unknown imbalances can introduce biases to both within- and between-study effect estimates. For example, in the extracted PsA RCTs, year of publication or country of study can convey different accessibilities based on the treatment's availability in a healthcare system at that time. However, a specific measured variable representing access issues was not available. Additionally, comorbidity distributions were not part of the study extraction list because they are often unreported in PsA studies or only severe comorbidities are reported as exclusion criteria. Nevertheless, clinical or more conceptual rather than quantitative judgment can support decision-making when there is no covariate measure available (e.g., regarding differences in study eligibility or disease indication). The greater awareness and transparent reporting of these study characteristics and methodologies will help support robust ITC to inform clinicians, patients and healthcare decision-makers when deciding courses of treatment among the large array of therapies for a given indication.
Conclusion
Using PsA RCTs as a case study, 18 eligible studies were available to conduct the NMA. Among the extracted studies were numerous violations of transitivity for the proposed network. Varying efforts, including reporting, were performed to identify potential effect modifiers that could introduce bias into the network. However, between-study heterogeneity is often underevaluated in published ITCs. More formal and comprehensive transitivity criteria were developed with the aim of informing the appropriateness and validity of future ITCs for clinical trials. Unbiased and informed decision-making is crucial for high-quality patient care.
•
An indirect treatment comparison (ITC) is used to compare two or more treatments when no direct, head-to-head study comparisons are available.
•
To provide appropriate comparisons, the treatment network should satisfy the transitivity assumption, that is, equal likelihood of treatment assignment for a given patient based on comparability of studies.
•
This case study of psoriatic arthritis (PsA) aimed to develop a criterion-based approach to evaluate challenges in satisfying the transitivity assumption and select appropriate studies for ITC. PsA was used as a case study to demonstrate study selection and network evaluation after performing a systematic literature review for ITC analysis.
•
We developed a framework to determine the plausibility of study comparisons and applied it to 18 RCTs, comparing apremilast with seven other treatments in PsA with no available direct comparisons.
•
Seven criteria were evaluated to include studies in the ITC: inclusion/exclusion criteria; clinical trial design and follow-up; baseline characteristics; disease severity subgroups; prior therapies; concomitant and extended-trial treatment and differences in placebo response.
•
To enable the generation of robust and reliable ITC estimates, several key patient and trial characteristics from the selected studies and examples of imbalanced baseline characteristics and other clinical trial design differences potentially introducing biases due to confounding were identified.
•
However, most studies were proven to contribute to a lack of transitivity in the network due to disparities in clinical trial design (including crossover, time of efficacy assessment and follow-up) and population differences (baseline disease characteristics, prior and concomitant medications and placebo response rates).
•
The inability to satisfy the transitivity assumption often results from multiple challenges. Transitivity assumptions must be evaluated thoroughly, as biases due to confounding are often underevaluated when performing ITC.
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: Supplementary Material
Author contributions
G Tremblay made substantial contributions to the conception and design of the work, analyzed and interpreted data and critically revised the article for important intellectual content. T Westley made substantial contributions to the design of the work, analyzed and interpreted data and drafted the article. A Forsythe and C Pelletier made substantial contributions to the conception and design of the work, interpreted data and critically revised the article for important intellectual content. A Briggs interpreted data and critically revised the article for important intellectual content. All authors approved the final version to be published and agreed to be accountable for all aspects of the work.
Financial & competing interests disclosure
This study was sponsored by Celgene Corporation. G Tremblay, T Westley and A Forsythe are employees of Purple Squirrel Economics. C Pelletier is an employee of Celgene Corporation. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
The authors would like to thank J Hearnden for her assistance in preparing the manuscript. The authors also received editorial support in the preparation of this manuscript from Peloton Advantage, LLC, an OPEN Health company, Parsippany, NJ, USA, sponsored by Celgene Corporation, Summit, NJ, USA. The authors, however, directed and are fully responsible for all content and editorial decisions for this manuscript.
Open access
This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
Data sharing statement
Celgene is committed to responsible and transparent sharing of clinical trial data with patients, healthcare practitioners and independent researchers for the purpose of improving scientific and medical knowledge as well as fostering innovative treatment approaches. For more information, please visit: https://www.celgene.com/research-development/clinical-trials/clinical-trials-data-sharing/.
Supplementary Material
File (suppl_data.docx)
- Download
- 31.24 KB
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
1.
Salanti G. Indirect and mixed-treatment comparison, network, or multiple-treatments meta-analysis: many names, many benefits, many concerns for the next generation evidence synthesis tool. Res. Synth. Methods 3(2), 80–97 (2012).
• Comprehensive review of indirect network comparisons that highlights important quality concerns in past designs. Definitions and descriptions of key terminology were applied within this report.
2.
Sutton A, Ades AE, Cooper N, Abrams K. Use of indirect and mixed treatment comparisons for technology assessment. Pharmacoeconomics 26(9), 753–767 (2008).
3.
The Airways Group: Cochrane Methods. Transitivity assumption (2014). https://airways.cochrane.org/sites/airways.cochrane.org/files/public/uploads/The%20transitivity%20assumption%20CJC.pdf
• Defines the transitivity assumption and highlights potential problems in network meta-analyses. These definitions and concepts were applied within this report.
4.
Hutton B, Salanti G, Caldwell DM et al. The PRISMA extension statement for reporting of systematic reviews incorporating network meta-analyses of health care interventions: checklist and explanations. Ann. Intern. Med. 162(11), 777–784 (2015).
5.
Statistical considerations in indirect comparisons and network meta-analysis. Cochrane Methods Training Event (PART 1) (2013). https://methods.cochrane.org/cmi/statistical-considerations-indirect-comparisons-and-network-meta-analysis
6.
Buckley F, Finckh A, Huizinga TW, Dejonckheere F, Jansen JP. Comparative efficacy of novel DMARDs as monotherapy and in combination with methotrexate in rheumatoid arthritis patients with inadequate response to conventional DMARDs: a network meta-analysis. J. Manag. Care Spec. Pharm. 21(5), 409–423 (2015).
7.
Jansen JP, Naci H. Is network meta-analysis as valid as standard pairwise meta-analysis? It all depends on the distribution of effect modifiers. BMC Med. 11(1), 159 (2013).
•• Discusses validity issues in indirect comparsions and demonstrates graphically how imbalances in effect modifiers or patient characteristics can introduce different biases into comparative effect estimates.
8.
Lunt M, Solomon D, Rothman K et al. Different methods of balancing covariates leading to different effect estimates in the presence of effect modification. Am. J. Epidemiol. 169(7), 909–917 (2009).
9.
Jansen JP, Fleurence R, Devine B et al. Interpreting indirect treatment comparisons and network meta-analysis for health-care decision making: report of the ISPOR Task Force on Indirect Treatment Comparisons Good Research Practices: Part 1. Value Health 14(4), 417–428 (2011).
10.
Park HM. Comparing group means: T-tests and one-way ANOVA using STATA, SAS, R and SPSS. The University Information Techology Services (UITS) Center for Statistical and Mathematical Computing, Indiana University, IN, USA (2009). http://www.indiana.edu/∼statmath/stat/all/ttes
11.
Thompson SG. Why sources of heterogeneity in meta-analysis should be investigated. BMJ 309(6965), 1351–1355 (1994).
12.
Caldwell DM. An overview of conducting systematic reviews with network meta-analysis. Syst. Rev. 3(1), 109 (2014).
13.
Catala-Lopez F, Tobias A, Cameron C, Moher D, Hutton B. Network meta-analysis for comparing treatment effects of multiple interventions: an introduction. Rheumatol. Int. 34(11), 1489–1496 (2014).
14.
Nikolakopoulou A, Chaimani A, Veroniki AA, Vasiliadis HS, Schmid CH, Salanti G. Characteristics of networks of interventions: a description of a database of 186 published networks. PLoS ONE 9(1), e86754 (2014).
15.
Cutolo M, Myerson GE, Fleischmann RM et al. A Phase III, randomized, controlled trial of apremilast in patients with psoriatic arthritis: results of the PALACE 2 trial. J. Rheumatol. 43(9), 1724–1734 (2016).
• Apremilast trial used as a reference point to which the rest of baseline patient characteristics across the different treatments were compared in the proposed indirect comparison network.
16.
Edwards CJ, Blanco FJ, Crowley J et al. Apremilast, an oral phosphodiesterase 4 inhibitor, in patients with psoriatic arthritis and current skin involvement: a Phase III, randomized, controlled trial (PALACE 3). Ann. Rheum. Dis. 75(6), 1065–1073 (2016).
• Apremilast trial used as a reference point to which the rest of baseline patient characteristics across the different treatments were compared in the proposed indirect comparison network.
17.
Ungprasert P, Thongprayoon C, Davis JM 3rd. Indirect comparisons of the efficacy of subsequent biological agents in patients with psoriatic arthritis with an inadequate response to tumor necrosis factor inhibitors: a meta-analysis. Clin. Rheumatol. 35(7), 1795–1803 (2016).
18.
Betts KA, Griffith J, Friedman A, Zhou ZY, Signorovitch JE, Ganguli A. An indirect comparison and cost per responder analysis of adalimumab, methotrexate and apremilast in the treatment of methotrexate-naive patients with psoriatic arthritis. Curr. Med. Res. Opin. 32(4), 721–729 (2016).
• Presents different network meta-analyses consisting of the same, recent trials within psoriatic arthritis and states that differences in patient characteristics across the indirect networks were potential limitations in study interpretation.
19.
Gharaibeh M, Xu Y, Lee J, Chitnis M, Collier D. Efficacy of biologics and new anti-inflammatory agents used in the treatment of active psoriatic arthritis: systematic literature review and network meta-analysis of the evidence. Arthritis Rheumatol. 69(Suppl. 10), Abstract 1556 (2017). https://acrabstracts.org/abstract/efficacy-of-biologics-and-new-anti-inflammatory-agents-used-in-the-treatment-of-active-psoriatic-arthritis-systematic-literature-review-and-network-meta-analysis-of-the-evidence/
• Presents different network meta-analyses consisting of the same, recent trials within psoriatic arthritis and states that differences in patient characteristics across the indirect networks were potential limitations in study interpretation.
20.
Strand V, Elaine Husni M, Betts KA et al. Network meta-analysis and cost per responder of targeted Immunomodulators in the treatment of active psoriatic arthritis. BMC Rheumatol. 2(1), 3 (2018).
21.
Christensen AW, Tarp S, Furst DE et al. Most trial eligibility criteria and patient baseline characteristics do not modify treatment effect in trials using targeted therapies for rheumatoid arthritis: a meta-epidemiological study. PLoS ONE 10(9), e0136982 (2015).
22.
Cochrane Handbook for Systematic Reviews of Interventions. Higgins JPT, Green S (Eds). The Cochrane Collaboration (2011). https://handbook-5-1.cochrane.org/
23.
Donegan S, Williamson P, Gamble C, Tudur-Smith C. Indirect comparisons: a review of reporting and methodological quality. PLoS ONE 5(11), e11054 (2010).
24.
Song F, Loke YK, Walsh T, Glenny A-M, Eastwood AJ, Altman DG. Methodological problems in the use of indirect comparisons for evaluating healthcare interventions: survey of published systematic reviews. BMJ 338, b1147 (2009).
•• Provides numerous examples of potential validity and interpretive issues with indirect comparison networks. Some issues identified were used to inform this report's proposed transitivity criteria.
25.
Yildiz A, Nikodem M, Vieta E, Correll CU, Baldessarini RJ. A network meta-analysis on comparative efficacy and all-cause discontinuation of antimanic treatments in acute bipolar mania. Psychol. Med. 45(2), 299–317 (2015).
26.
Tonin FS, Rotta I, Mendes AM, Pontarolo R. Network meta-analysis: a technique to gather evidence from direct and indirect comparisons. Pharm. Pract. 15(1), 943 (2017).
27.
Chaimani A. Graphical tools for network meta-analysis in STATA. PLoS ONE 8(10), e76654 (2013).
28.
Phillippo DM, Ades AE, Dias S et al. NICE DSU Technical Support Document 18: methods for population-adjusted indirect comparisons in submissions to NICE. NICE (2019). http://nicedsu.org.uk/ 695 wp-content/uploads/2018/08/Population-adjustment-TSD-FINALref-rerun.pdf
29.
Rothman KJ, Greenland S, Walker AM. Concepts of interaction. Am. J. Epidemiol. 112(4), 467–470 (1980).
30.
Puhan MA, Schunemann HJ, Murad MH et al. A GRADE Working Group approach for rating the quality of treatment effect estimates from network meta-analysis. BMJ 349, 5630 (2014).
31.
McInnes IB, Mease PJ, Kirkham B et al. Secukinumab, a human anti-interleukin-17A monoclonal antibody, in patients with psoriatic arthritis (FUTURE 2): a randomized, double-blind, placebo-controlled, Phase III trial. Lancet 386(9999), 1137–1146 (2015).
32.
Ritchlin C, Rahman P, Kavanaugh A et al. Efficacy and safety of the anti-IL-12/23 p40 monoclonal antibody, ustekinumab, in patients with active psoriatic arthritis despite conventional non-biological and biological anti-tumour necrosis factor therapy: 6-month and 1-year results of the Phase III, multicentre, double-blind, placebo-controlled, randomized PSUMMIT 2 trial. Ann. Rheum. Dis. 73(6), 990–999 (2014).
33.
Kavanaugh A, Mease PJ, Gomez-Reino JJ et al. Treatment of psoriatic arthritis in a Phase III randomized, placebo-controlled trial with apremilast, an oral phosphodiesterase 4 inhibitor. Ann. Rheum. Dis. 73(6), 1020–1026 (2014).
• Apremilast trial used as a reference point to which the rest of patient characteristics across the different treatments were compared in the proposed indirect comparison network.
34.
Schett G, Wollenhaupt J, Papp K et al. Oral apremilast in the treatment of active psoriatic arthritis: results of a multicenter, randomized, double-blind, placebo-controlled study. Arthritis Rheumatol. 64(10), 3156–3167 (2012).
• Apremilast trial used as a reference point to which the rest of patient characteristics across the different treatments were compared in the proposed indirect comparison network.
35.
McInnes IB, Kavanaugh A, Gottlieb AB et al. Efficacy and safety of ustekinumab in patients with active psoriatic arthritis: 1 year results of the Phase III, multicentre, double-blind, placebo-controlled PSUMMIT 1 trial. Lancet 382(9894), 780–789 (2013).
36.
Mease PJ, Fleischmann R, Deodhar AA et al. Effect of certolizumab pegol on signs and symptoms in patients with psoriatic arthritis: 24-week results of a Phase III double-blind randomized placebo-controlled study (RAPID-PsA). Ann. Rheum. Dis. 73(1), 48–55 (2014).
37.
Genovese MC, Mease PJ, Thomson GT et al. Safety and efficacy of adalimumab in treatment of patients with psoriatic arthritis who had failed disease modifying antirheumatic drug therapy. J. Rheumatol. 34(5), 1040–1050 (2007).
38.
Kavanaugh A, McInnes I, Mease P et al. Golimumab, a new human tumor necrosis factor alpha antibody, administered every four weeks as a subcutaneous injection in psoriatic arthritis: Twenty-four-week efficacy and safety results of a randomized, placebo-controlled study. Arthritis Rheumatol. 60(4), 976–986 (2009).
39.
Mease PJ, Gladman DD, Ritchlin CT et al. Adalimumab for the treatment of patients with moderately to severely active psoriatic arthritis: results of a double-blind, randomized, placebo-controlled trial. Arthritis Rheumatol. 52(10), 3279–3289 (2005).
40.
Mease PJ, Goffe BS, Metz J, Vanderstoep A, Finck B, Burge DJ. Etanercept in the treatment of psoriatic arthritis and psoriasis: a randomized trial. Lancet 356(9227), 385–390 (2000).
41.
Mease PJ, Kivitz AJ, Burch FX et al. Etanercept treatment of psoriatic arthritis: safety, efficacy, and effect on disease progression. Arthritis Rheumatol. 50(7), 2264–2272 (2004).
42.
Antoni C, Krueger GG, De Vlam K et al. Infliximab improves signs and symptoms of psoriatic arthritis: results of the IMPACT 2 trial. Ann. Rheum. Dis. 64(8), 1150–1157 (2005).
43.
Gottlieb A, Menter A, Mendelsohn A et al. Ustekinumab, a human interleukin 12/23 monoclonal antibody, for psoriatic arthritis: randomized, double-blind, placebo-controlled, crossover trial. Lancet 373(9664), 633–640 (2009).
44.
McInnes IB, Sieper J, Braun J et al. Efficacy and safety of secukinumab, a fully human anti-interleukin-17A monoclonal antibody, in patients with moderate-to-severe psoriatic arthritis: a 24-week, randomized, double-blind, placebo-controlled, Phase II proof-of-concept trial. Ann. Rheum. Dis. 73, 349–356 (2014).
45.
Antoni CE, Kavanaugh A, Kirkham B et al. Sustained benefits of infliximab therapy for dermatologic and articular manifestations of psoriatic arthritis: results from the infliximab multinational psoriatic arthritis controlled trial (IMPACT). Arthritis Rheumatol. 52(4), 1227–1236 (2005).
46.
Atteno M, Peluso R, Costa L et al. Comparison of effectiveness and safety of infliximab, etanercept, and adalimumab in psoriatic arthritis patients who experienced an inadequate response to previous disease-modifying antirheumatic drugs. Clin. Rheumatol. 29(4), 399–403 (2010).
47.
Kingsley GH, Kowalczyk A, Taylor H et al. A randomized placebo-controlled trial of methotrexate in psoriatic arthritis. Rheumatology (Oxford) 51(8), 1368–1377 (2012).
48.
Willkens RF, Williams HJ, Ward JR et al. Randomized, double-blind, placebo controlled trial of low-dose pulse methotrexate in psoriatic arthritis. Arthritis Rheumatol. 27(4), 376–381 (1984).
49.
Wells A, Edwards C, Adebajo AO et al. PALACE 4, a Phase III, randomized, controlled trial of apremilast, an oral phosphodiesterase 4 inhibitor, for treatment of psoriatic arthritis: long-term (52-week) improvements in physical function [abstract SAT0382]. Ann. Rheumatol. 73(Suppl. 2), 732 (2014).
50.
Chaimani A, Caldwell DM, Li T, Higgins JPT, Salanti G. Additional considerations are required when preparing a protocol for a systematic review with multiple interventions. J. Clin. Epidemiol. 83, 65–74 (2017).
51.
Ballegaard CJ, Jørgensen TS, Skougaard TS et al. Assessing the importance of trial characteristics as contextual factors when evaluating targeted therapies in patients with psoriatic disease: protocol for an exploratory systematic review and meta-research project. The Parker Institute (2016). http://www.parkerinst.dk/sites/default/files/study_protocol_3.pdf
52.
Briggs AM, March L, Lassere M et al. Baseline comorbidities in a population-based cohort of rheumatoid arthritis patients receiving biological therapy: data from the Australian rheumatology association database. Int. J. Rheum. 2009, 861481 (2009).
53.
Latimer NRA, Abrams KR. Adjusting survival time estimates in the presence of treatment switching. NICE DSU Technical Support Document 16. National Institute for Health and Care Excellence (NICE), London, UK (2014). http://nicedsu.org.uk/wp-content/uploads/2016/03/TSD16_Treatment_Switching.pdf
54.
Toh S, Li L, Harrold LR et al. Comparative safety of infliximab and etanercept on the risk of serious infections: does the association vary by patient characteristics? Pharmacoepidemiol. Drug Saf. 21(5), 524–534 (2012).
55.
Thorlund K, Druyts E, Avina-Zubieta JA, Wu P, Mills EJ. Why the findings of published multiple treatment comparison meta-analyses of biologic treatments for rheumatoid arthritis are different: an overview of recurrent methodological shortcomings. Ann. Rheum. Dis. 72(9), 1524–1535 (2013).
56.
Saad ED, Buyse M. Statistical controversies in clinical research: end points other than overall survival are vital for regulatory approval of anticancer agents. Ann. Oncol. 27(3), 373–378 (2016).
57.
Corbett M, Chehadah F, Biwas M et al. Certolizumab pegol and secukinumab for treating active psoriatic arthritis following inadequate response to disease modifying anti-rheumatic drugs: a systematic review and economic evaluation. Health Technol. Assess. 21(56), 1–326 (2017).
58.
Van SS, Diels J, Van LJ, Hemels M. Network meta-analysis with baseline risk adjustment to assess the relative efficacy of ustekinumab in adult patients with active psoriatic arthritis. Value Health 17(7), A373 (2014).
59.
Gladman DD, Thavaneswaran A, Chandran V, Cook RJ. Do patients with psoriatic arthritis who present early fare better than those presenting later in the disease? Ann. Rheum. Dis. 70(12), 2152–2154 (2011).
60.
Theander E, Husmark T, Alenius GM et al. Early psoriatic arthritis: short symptom duration, male gender and preserved physical functioning at presentation predict favourable outcome at 5-year follow-up. Results from the Swedish Early Psoriatic Arthritis Register (SwePsA). Ann. Rheum. Dis. 73(2), 407–413 (2014).
61.
Tillett W, Jadon D, Shaddick G et al. Smoking and delay to diagnosis are associated with poorer functional outcome in psoriatic arthritis. Ann. Rheum. Dis. 72(8), 1358–1361 (2013).
62.
McInnes IB, Nash P, Ritchlin C et al. Secukinumab for the treatment of psoriatic arthritis: comparative effectiveness results versus licensed biologics and apremilast from a network meta-analysis [abstract THU0437]. Ann. Rheum. Dis. 75(Suppl. 2), 348–349 (2016).
Information & Authors
Information
Published In
Pages: 1265 - 1298
PubMed: 31774340
Copyright
© 2019 Gabriel Tremblay. This work is licensed under the Attribution-NonCommercial-NoDerivatives 4.0 Unported License
History
Received: 23 May 2019
Accepted: 20 August 2019
Published online: 5 September 2019
Keywords:
Topics
Authors
Metrics & Citations
Metrics
Article Usage
Article usage data only available from February 2023. Historical article usage data, showing the number of article downloads, is available upon request.
Citations
How to Cite
A criterion-based approach to systematic and transparent comparative effectiveness: a case study in psoriatic arthritis. (2019) Journal of Comparative Effectiveness Research. DOI: 10.2217/cer-2019-0064
Export citation
Select the citation format you wish to export for this article or chapter.
Citing Literature
- Maria De Santis, Sara Martínez Breijo, Paul Robinson, Camille Capone, Katie Pascoe, Suzy Van Sanden, Mahmoud Hashim, Marco Trevisan, Caitlin Daly, Friso Reitsma, Sophie van Beekhuizen, Haoyao Ruan, Bart Heeg, Elena Verzoni, Feasibility of Indirect Treatment Comparisons Between Niraparib Plus Abiraterone Acetate and Other First-Line Poly ADP-Ribose Polymerase Inhibitor Treatment Regimens for Patients with BRCA1/2 Mutation-Positive Metastatic Castration-Resistant Prostate Cancer, Advances in Therapy, 10.1007/s12325-024-02918-6, 41, 8, (3039-3058), (2024).