Study protocol for the dabigatran, apixaban, rivaroxaban, edoxaban, warfarin comparative effectiveness research study
Abstract
Since 2010, four oral anticoagulants have been approved for marketing in addition to warfarin for treatment of thromboembolic disease. Limited head-to-head data exist comparing these treatments, leaving patients and clinicians with little guidance for selecting a strategy that balances recurrence reduction with bleeding risk. In the dabigatran, apixaban, rivaroxban, edoxaban and warfarin comparative effectiveness research study, we compare all five currently available oral anticoagulant agents for the extended treatment of deep venous thrombosis and pulmonary embolism, as well as no extended treatment, and evaluate whether results differ in specific sub-populations. As our population includes Medicare novel anticoagulant users and large numbers of commercially insured and Medicaid patients, our results will likely be transportable to the majority of US patients experiencing a DVT or pulmonary embolism. Clinical Trials registration: NCT03271450.
Each year, approximately 500,000 Americans experience venous thromboembolic disease, including deep venous thrombosis (DVT) and pulmonary embolism (PE) [1,2]. For more than five decades, the vitamin K antagonist warfarin was essentially the only oral anticoagulant available for management of venous thromboembolic disease. Between 2010 and 2015, four new oral anticoagulants (NOACs) were approved for marketing: the direct thrombin inhibitor, dabigatran and the direct factor Xa inhibitors, rivaroxaban, apixaban and edoxaban.
Several randomized trials have compared the safety and efficacy of the NOACs to warfarin for the initial management of venous thromboembolic disease; however, little data exist comparing the effectiveness and safety of these drugs for extended treatment of DVT and PE. In one placebo-controlled randomized trial, extended treatment with dabigatran was highly effective in reducing recurrent venous thromboembolism compared with no extended anticoagulation, but patients in the dabigatran group were nearly three-times more likely to experience major or clinically relevant bleeding [3]. In another randomized trial comparing extended treatment with dabigatran to extended treatment with warfarin, the rate of recurrent venous thromboembolism was higher in the dabigatran group compared with the warfarin group (hazard ratio [HR]: 1.44; 95% CI: 0.78–2.64), but fewer patients in the dabigatran group experienced major or clinically relevant bleeding (HR: 0.54; 95% CI: 0.41–0.71) [3]. The EINSTEIN trial found that extended treatment with rivaroxaban compared with placebo substantially decreased recurrent venous thromboembolism (HR: 0.18; 95% CI: 0.09–0.39), however low event numbers limited evaluation of bleeding risk; the more recent EINSTEIN CHOICE trial established the effectiveness of both treatment and therapeutic doses of rivaroxaban compared with aspirin, with no compensatory increase in bleeding [4,5]. Finally, a trial of extended apixaban versus placebo use found substantially reduced risk of recurrent venous thromboembolism or death without increased risk of major bleeding [4,6].
While these studies are important for establishing the efficacy of NOACs in the extended treatment of venous thromboembolic disease, they provide very limited head-to-head data, leaving patients, clinicians and other stakeholders with little guidance for selecting the best strategy that balances recurrence reduction with risk of bleeding. Clinical guidelines recommend extended treatment beyond 3 months for patients in certain clinical scenarios, such as when the DVT is unprovoked, however they do not provide insight on which oral anticoagulants should be used for the extended treatment of DVT and PE nor on the optimal duration of therapy [7]. The recently published 2016 Chest Guidelines for antithrombotic therapy highlight the limited available evidence and need for a comparative safety and effectiveness study [8].
In the dabigatran, apixaban, rivaroxaban, edoxaban (DARE) warfarin comparative effectiveness research study, we compare all five currently available oral anticoagulant agents for the extended treatment of DVT and PE, as well as no extended treatment. The study compares these options with respect to effectiveness in reducing venous thromboembolism recurrence risk and safety in reducing intracranial bleed, gastrointestinal bleeding and other major bleeding risk, as well as the relative impact on all-cause mortality. This study will also determine whether treatment heterogeneity exists for specific populations, such as older patients or those with renal dysfunction. By comparing all five options, this study will directly address the critical clinical tradeoffs patients and their healthcare providers make when selecting an anticoagulant for the extended treatment of DVT and PE and provide valuable guidance for clinical practice. In this report, we describe the design of the DARE Warfarin CER Study.
Study design
Overview
The primary aim of this study is to compare treatment for at least 90, 180, 270 and 360 days across five currently available oral anticoagulants over a study period from 2009 to 2020: warfarin, dabigatran, apixaban, rivaroxaban and edoxaban; the study will also compare extended treatment to earlier discontinuation of anticoagulant therapy. Each of the treatments has been approved by the US FDA for the treatment of DVT and PE and these approvals have been supported by one or more large randomized trials establishing the efficacy of each drug in reducing recurrence of DVT and PE [9–12].
The study includes three additional aims to support conclusions from the primary aim: (Aim 2) to conduct validation of key study variables and subgroup analyses within a subset of patients for whom claims and electronic health record (EHR) data are available; (Aim 3) to examine heterogeneity in treatment effects across multiple patient factors; (Aim 4) to calibrate study results to those of available randomized trials. Final design and analysis decisions will be made following review of available sample size and feedback received from the Study Advisory Committee. Both of these inputs are described below.
Stakeholder engagement
The Study Advisory Committee will be composed of multiple patients and stakeholders that will be highly engaged throughout the research process to ensure that the study yields results that are maximally useful for improving patient-centered outcomes. The committee was convened in the development of the study proposal, and provided valuable input into the study design at that time. During the granting period, the committee will meet regularly to discuss design considerations, such as subgroups to study, outcome definitions and statistical considerations that support the translational aims of the grant. The Study Advisory Committee will also have an active role in disseminating study results. Together with the committee, we will identify stakeholders and patients, including national or regional organizations that represent, at minimum patients and/or families with lived experience, relevant clinicians, payers and health plans. In particular, we aim to work with the North American Thrombosis Forum, a multidisciplinary organization with the mission of improving patient care through the advancement of thrombosis education, to disseminate results to patients and other stakeholders.
Study population
The study population will comprise individuals with an initial episode of DVT or PE and no prior oral anticoagulant use who complete 3 months of treatment with an oral anticoagulant. Patients will be identified in two large electronic administrative claims healthcare databases (described below). Combined, these databases include information for more than 215 million Americans. Validation sub-cohort analyses (Aim 2) will consist of patients receiving care in eastern Massachusetts for whom we have EHR data linked to Medicare claims data available, and patients in the Truven commercial database with laboratory data available.
Patient inclusion & exclusion criteria
Included patients will be those in our databases with a first inpatient diagnosis of DVT or PE following a period of at least 1 year with no evidence of a prior DVT or PE and no use of an oral anticoagulant. Initial claims-based screening definitions for DVT or PE will be based on International Classification of Disease (ICD) codes (Table 1). The patients will be eligible for inclusion if they fill a prescription for an oral anticoagulant at an indicated dose (warfarin, dabigatran 150 mg, rivaroxaban 15 or 20 mg, apixaban 2.5, 5 or 10 mg and edoxaban 30 or 60 mg) within 7 days of the initial DVT or PE event and continuously use an anticoagulant for at least 90 days, defined as no gaps in therapy greater than 7 days. Patients experiencing a study outcome between discharge for the index DVT/PE event and 90 days after will be excluded. The overall study design is summarized in Figure 1 and cohort flow in Figure 2.
| Outcome | ICD-9 codes | Validation | Ref. |
|---|---|---|---|
| Deep venous thromboembolism or pulmonary embolism | 415.x: acute pulmonary heart disease 451.x: phlebitis and thrombophlebitis of deep veins of upper extremities 453.x: other venous embolism and thrombosis | PPV: 65–95% | [33] |
| Major extracranial bleed | Gastrointestinal bleed + 423.0x: hemopericardium 599.7x: hematuria 719.11: hemarthrosis, shoulder region 784.7x: epistaxis 784.8x: hemorrhage from throat 786.3x: hemoptysis | – | – |
| Gastrointestinal bleed | 455.2x: internal hemorrhoids with other complication 455.5x: external hemorrhoids with other complication 455.8x: unspecified hemorrhoids with other complication 456.0x: esophageal varices with bleeding 456.20: esophageal varices in diseases classified elsewhere, with bleeding 530.7x: gastroesophageal laceration-hemorrhage syndrome 530.82: esophageal hemorrhage 531.0x - 531.6x: gastric ulcer (excluding without mention of hemorrhage or perforation) 532.0x - 532.6x: duodenal ulcer (excluding without mention of hemorrhage or perforation) 533.0x - 533.6x: peptic ulcer site unspecified (excluding without mention of hemorrhage or perforation) 534.0x - 534.6x: gastrojejunal ulcer (excluding without mention of hemorrhage or perforation) 535.01 - 535.61: gastritis (excluding acute gastritis without mention of hemorrhage; duodenitis; eosinophilic gastritis) 537.83: angiodysplasia of stomach and duodenum with hemorrhage 562.02, 562.03, 562.12, 562.13: diverticula of intestine (excluding without mention of hemorrhage) 568.81: hemoperitoneum (nontraumatic) 569.3x: hemorrhage of rectum and anus 569.85: angiodysplasia of intestine with hemorrhage 578.xx: gastrointestinal hemorrhage | PPV: 78.7–94.0% | [34] |
| Intracranial bleed | 430.x: subarachnoid hemorrhage 431.x: intracerebral hemorrhage 432.x: other and unspecified intracranial hemorrhage 852.0x: subarachnoid subdural and extradural hemorrhage following injury 852.2x: subdural hemorrhage following injury without mention of open intracranial wound 852.4x: extradural hemorrhage without mention of open intracranial wound 853.0x: other and unspecified intracranial hemorrhage following injury | PPV: 77% | [35] |
ICD: International Classification of Disease; PPV: Positive predictive value.
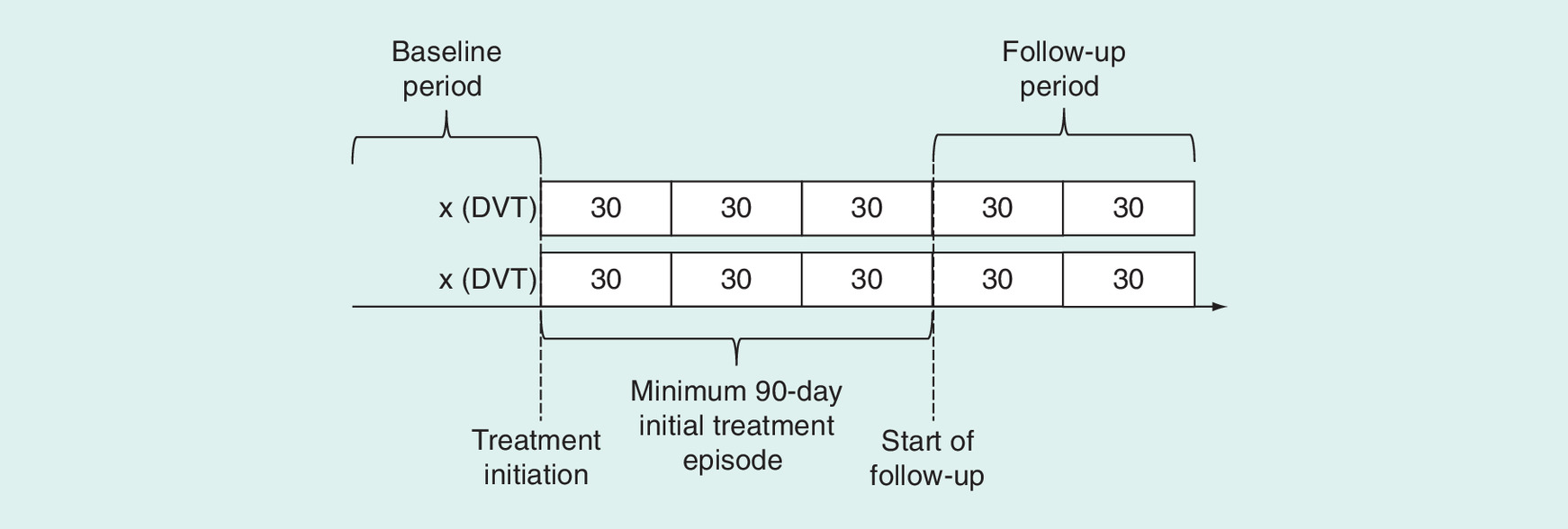
Figure 1. Study design.
DVT: Deep venous thrombosis.
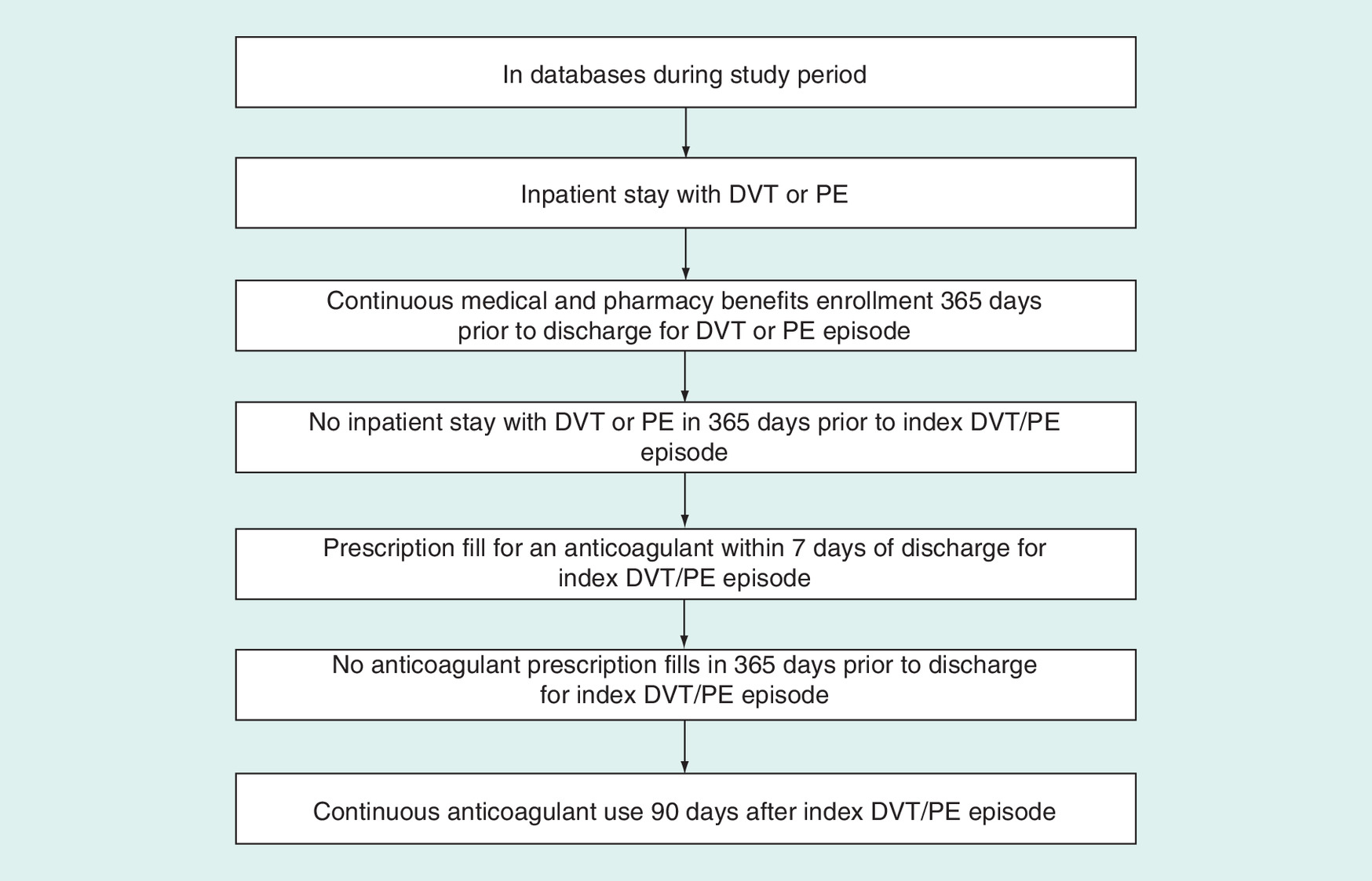
Figure 2. Flowchart of study inclusion criteria.
DVT: Deep venous thrombosis; PE: Pulmonary embolism.
Exposure
The patients will be defined as being ‘on therapy’ at 3 months if they have continuous medication supply available at the 90-day mark. The patients will be defined as continuers if they refill an anticoagulant within 14 days after the end of days supply of the fill spanning day 90; patients without an anticoagulant in this period will be classified as discontinuers. The index date for both continuers and discontinuers is the end of the 14-day period after the end of days supply. The patients must additionally be event free and retain medical and pharmacy benefits through the index date. These procedures will be repeated to define exposure at 180, 270, and 360 days among the subsets of patients who continue therapy through these dates.
Therapy continuers will be classified into individual anticoagulant groups. There are several options for assigning exposure to the subset of patients who switch anticoagulant therapy prior to the start of follow-up. We will consider assigning the patient to the anticoagulant they are taking in their first fill after discharge for DVT/PE; assigning the patient to the anticoagulant they are taking at the start of follow-up; assigning patients with a switch to a separate exposure category. We will evaluate these three options with regard to study validity (bias reduction) and available sample size and assign one as the primary analysis and the two others as sensitivity analyses.
End points
The primary study outcome is major bleeding, defined as intracranial bleed, gastrointestinal bleed or other major bleeding episode. Secondary study outcomes include DVT or PE recurrence, defined as a new episode of either condition during follow-up; composite of the primary or first secondary outcome (safety and effectiveness outcomes); and composite of the primary or first secondary outcome including all-cause mortality. Outcomes will be defined using claim-based algorithms that have been shown to have high-positive predictive values (Table 1).
Covariates
We will measure a large number of potential confounders. These include socio-demographic characteristics (e.g., age, sex, race where available, geographic region and receipt of low-income subsidy for Medicare beneficiaries) risk factors for bleeding (e.g., prior hospitalized bleed, gastric ulcer disorder, coagulation defects), risk factors for thromboembolism and other thrombotic events (e.g., prior ischemic stroke, transient ischemic attack, acute coronary syndrome, surgery or trauma, infection), prescription drug use (e.g., antiplatelet, statin, anti-inflammatory medications) and proxy measures of health status and frailty (e.g., medication burden, prior hospitalizations, combined comorbidity score) [13]. Covariates will be measured in the period 365 days prior to the index DVT/PE episode date through the index date, for each of the four minimum-exposure durations.
Data collection & analysis
Data sources
Our study will include two data sources: Fee-for-service Medicare claims data from 2009 to 2019 and Truven MarketScan commercial and Medicaid data from 2009 to 2020. The Truven MarketScan Commercial Claims and Encounters database represents 141.8 million privately insured individuals and captures patient-level de-identified data from inpatient and outpatient visits and pharmacy claims of multiple insurance plans. The Truven MarketScan Multi-State Medicaid database contains administrative claims data for 19.9 million Medicaid enrollees from multiple states, including inpatient, outpatient and pharmacy services.
Aim 2 will be conducted in the subset of Medicare beneficiaries who receive care at Brigham and Women's Hospital and Massachusetts General Hospital along with a number of smaller hospitals and centers as well as the Partners Community Physician Organization, a large network of outpatient physician practices in the Boston area. For these patients, we will link Medicare claims data to EHR data from the Partners Healthcare EHR data system, which contain information on disease severity, smoking status, body mass index, laboratory and radiology test results, cancer grade and stage and drug allergy histories [14]. The total size of this cohort is estimated to be 1 million patients. For Aim 2, we will additionally access the MarketScan Lab Supplemental database, which represents 4.4 million privately insured individuals with at least one recorded laboratory value, with administrative claims from inpatient, outpatient and pharmacy services supplemented by laboratory results.
Statistical analysis
The main analysis (Aim 1) consists of comparison of the five oral anticoagulants, as well as a comparison to no extended anticoagulant treatment. To account for the nonrandom allocation of patients to exposure groups in our study, we will construct propensity scores that summarize all potential confounders. We will use propensity score matching weights, a weighting analog to matching recently extended to more than two treatments, in outcome regression models [15,16]. We will use standard diagnostics for inverse probability of treatment weighting, including examining mean weights across treatment groups and assessing for extreme weights; if extreme weights are observed, we will consider standard approaches to address them, including use of stabilized weights and weight truncation. We will also examine overlap in propensity score distributions across treatment groups and we will assess covariate balance across treatment groups in the weighted population using standardized differences [2].
In addition to prespecified covariates, the propensity score will consist of variables selected by the high-dimensional propensity score algorithm to enrich the models with variables that might be confounders that were not considered among the predefined covariates. The approach identifies and prioritizes potential confounders by estimating the confounding potential for a large number (usually hundreds or thousands) of codes in the database by calculating the association between each code and the exposure, the association between each code and the outcome, and the prevalence of each code and enters these into a bias formula. Both simulation and empirical studies have found that high-dimensional propensity scores perform as well or better than propensity scores that include only investigator-specified variables [17–19].
Outcomes for each minimum exposure duration (continuation beyond 90 days; continuation beyond 180 days; continuation beyond 270 days; continuation beyond 360 days) will be modeled using Cox proportional hazards models. The primary analysis will be an ‘as treated’ approach, where patients are censored at treatment discontinuation or switch in addition to loss of insurance eligibility or death. In a secondary analysis, we will follow patients in an intention-to-treat fashion.
We will update the data periodically during the course of the study to maximize sample size at the end of the funding period. We will use sequential testing strategies, based on the maximum sequential probability ratio test, that have become standard tools for vaccine and drug safety monitoring by the CDC and the FDA, to maintain type I error rates [20–24]. As we are using routinely collected data, the study will continue should we reach statistical significance for any of our comparisons, however we will report interim results to demonstrate at what sample size statistical significance was met. The sequential testing algorithms for each outcome will account for the five data updates we will receive during the study period by ensuring a global type I error rate of 0.05. A key strength of the maximum sequential probability ratio test-based sequential testing methods is that they are highly flexible by accommodating any data updating and enrollment schedule.
Aim 2 seeks to validate key study variables to refine claim-based variable definitions. First, we will validate and refine the cohort identification definition for DVT and PE. To do so, we will first use the EHR data to validate each cohort-defining DVT or PE event identified in the claims for patients in the Medicare-EHR-linked subset. We will then randomly split the confirmed and unconfirmed cases into training and testing sets. In the training set of confirmed and unconfirmed cases, we will use recursive partitioning, a tree-based discrimination method, to select claims characteristics that best distinguish, in a sequential manner, medical record-confirmed cases of incident DVT or PE [25,26].
Second, we will use EHR data to assess and correct for residual confounding in the main claim-based analyses. We will use the availability of these variables such as BMI, smoking and aspirin use in the EHR data to evaluate the extent to which there are differences among these variables across treatment groups both before and after propensity score weighting. If no imbalances are observed or if weighting removes all imbalances that are present before weighting, then we can be reasonably assured that the main propensity score weighting approach sufficiently addresses confounding. If imbalances remain after weighting, we will repeat analyses of effect estimation within the validation subset with and without the EHR-only variables in the propensity score model. If inclusion of these variables in the propensity score does not materially change the results, then we can again be reasonably assured that the main propensity score weighting approach sufficiently addresses confounding. If inclusion of these variables changes the results, then we will use propensity score calibration to correct our main study results for this unmeasured confounding.
Third, we will use these data to examine the impact of incorporating International Normalized ratio data for warfarin patients in the analysis. Specifically, we will assess time in therapeutic range during the initial anticoagulant treatment episode for each warfarin initiator using outpatient INR test results. We will then stratify comparisons to warfarin by whether the warfarin patients had high or low time in therapeutic range. In addition to updating our main findings from Aim 1, as described above, we will also conduct refined subgroup analyses.
To address ‘missingness’ of EHR and laboratory data in the validation subsets, we will examine the frequency and patterns of missingness and use standard statistical imputation methods, including Markov Chain Monte Carlo and Bayesian imputation approaches, to impute missing observations. By using different imputation methods to compare the results of analyses that incorporate EHR and/or laboratory data from the validation subset to those that do not, we will be able to examine the sensitivity of inferences to missing data methods and assumptions and incorporate them into interpretation.
In Aim 3, we will evaluate treatment effect heterogeneity using two approaches. First, we will pre-define subgroups, in particular age, sex, renal dysfunction and trauma or surgery. The final list of subgroups will include specific input from the Study Advisory Committee and will be finalized prior to any analyses. Where sufficient numbers of patients exposed to each treatment are available within the subgroups of interest, we will refit the propensity score models within each subgroup. When too few patients are available in a given subgroup to support propensity score estimation, the subgroups will be formed based on the variables included in the propensity score model, but separate models will not be fit within these subgroups.
Second, we will estimate disease risk scores for each outcome of interest and examine relative treatment effectiveness and safety across predicted probabilities of outcome risk. Disease risk scores are outcome prediction models that can include may potential predictors and can be used to estimate patients’ baseline outcome risk at the start of follow-up. We will use a historical cohort of warfarin initiators to develop each disease risk score model [27].
In Aim 4, we will calibrate study results to findings from randomized clinical trials for particular comparisons for which trial results are available. First, we will standardize our observational study results to the characteristics of the relevant trials. Standardization is a well established epidemiologic approach to generalizing the results of one study to a different patient population that can have different distributions of treatment effect modifiers. Similarity in standardized study and randomized trial results will provide reassurance of the validity and robustness of our observational study. If differences in results are observed, sources of discrepancies will be investigated in the validation subset in Aim 2, where additional assessment of unmeasured confounding will be performed.
Sensitivity analyses
For Aim 1, we will conduct two principal sensitivity analyses. To evaluate the extent to which residual confounding might influence our primary analysis results, we will conduct an instrumental variable (IV) analysis using the drug that the prescriber chose for the most recent patient as the IV, first confirming that this instrument reasonably meets the required IV assumptions [28]. Similar results to our main analysis will provide reassurance that the main results using propensity score weighting are not substantially confounded. Second, we will conduct analyses using a negative control outcome of cancer, which has been previously used in a study of statin use [29,30]. Negative control outcomes are outcomes known or hypothesized to be causally unassociated with the exposure of interest and thus can be used to test for residual confounding in using the same model as the main analysis. We will also consider using fracture, pneumonia and vaccination incidence as alternate negative control outcomes, defined a priori. In both the IV and negative control outcome analyses, if differences in results are observed, sources of discrepancies will be investigated in the validation subset in Aim 2, where additional assessment of unmeasured confounding will be performed.
Additional prespecified sensitivity analyses will test the robustness of our findings to various analytical decisions. We will re-run the analyses varying specifications around time between medication filling, for instance the time to index fill and the maximum allowable gap between fills. Additionally, we will perform quantitative bias analyses to assess the robustness of our study results to misclassification of exposures, outcomes and confounders, including unmeasured confounding [31,32]. Quantitative bias analysis methods quantify residual bias in effect estimation by modeling structural assumptions about confounding and misclassification mechanisms that can cause bias. They also assess the impact of these potential mechanisms on changes in study results.
Discussion
Clinical guidance on the comparative safety and effectiveness of long-term anticoagulant therapy is urgently needed. Patients and providers today face a choice among many treatments for the long-term reduction of venous thromboembolism recurrence after DVT or PE, and must also weigh the risk of bleeding with each of these therapies. There is currently insufficient data available from randomized trials to provide appropriate recommendations to patients and providers. The DARE Warfarin CER Study aims to close this knowledge gap and provide robust and highly implementable comparative effectiveness results for the main available anticoagulant treatment options.
This study has several important strengths. As our population includes all Medicare novel anticoagulant users and large numbers of commercially insured and Medicaid patients, our results are likely transportable to the majority of US patients discharged from the hospital after a DVT or PE. Moreover, our study evaluates treatment effect heterogeneity in important subgroups, and can therefore provide relevant information to these populations. The patients in our study are followed for up to 10 years, offering long-term assessment that is rarely available in studies involving prospective data collection. Additionally, the study incorporates rich, population-based clinical data for a subset of the study population. Combined with innovative approaches to dealing with misclassification bias or confounding that can arise in observational studies, these data will be used to evaluate and minimize potential bias. Our statistical analysis includes several key validation steps and methodological approaches to assist in optimally controlling for confounding while also exploring and quantifying the degree of potential residual confounding.
Our study has important advantages over a randomized trial design. The trials that have been completed to date provide little evidence about the comparative bleeding safety of the NOACs and warfarin in extended treatment of venous thromboembolic disease. Moreover, it is unlikely that a randomized trial would be large enough to compare major bleeding across all oral anticoagulants. Large observational studies such as this one are particularly well suited to study rare safety events. While possible in theory, a randomized trial is not feasible to answer the question that this study will address, namely the long-term comparative safety and effectiveness of extended treatment with all oral anticoagulants in a very large cohort of patients treated in the ‘real world’, including all Medicare patients and with sufficient sample size to evaluate many subgroup analyses and long-term, uncommon outcomes. Nonetheless, our study, as with all observational studies, is subject to bias, which we have attempted to mitigate in the design considerations detailed in the report. Administrative claims data may not capture important predictors of treatment that are also correlates of the study outcome, resulting in residual confounding by these unmeasured characteristics. Of note, claims data have limited information on reasons for treatment discontinuation, which may bias our ‘as treated’ effect estimates if reasons for discontinuation differ by treatment group. Consideration of discontinuation patterns as well as comparison of as treated and intention-to-treat effect estimates will be critical to evaluating and mitigating these concerns.
Results from this study will have an immediate and important impact on treatment decisions in clinical practice. To that end, in addition to formal publication of study results, we will develop several briefs summarizing the study results and implications to patients, clinicians and other decision-makers. These materials will be developed and disseminated in collaboration with stakeholders.
Conclusion
The comparative safety and effectiveness of long-term oral anticoagulant therapy in real-world settings has not been evaluated. The DARE Warfarin CER study aims to provide comprehensive evidence to guide treatment decisions of therapy choice and treatment duration.
Each year, approximately 500,000 Americans experience venous thromboembolic disease, including deep venous thrombosis (DVT) and pulmonary embolism (PE). Little data exist comparing the effectiveness and safety of available anticoagulants for extended treatment of DVT and PE. The dabigatran, apixaban, rivaroxaban, edoxaban, warfarin comparative effectiveness research study compares five currently available oral anticoagulant agents for the extended treatment of DVT and PE, as well as no extended treatment with respect to effectiveness and safety. This study will directly address the critical clinical tradeoffs patients and their healthcare providers make when selecting an anticoagulant for the extended treatment of DVT and PE and provide valuable guidance for clinical practice.
Study design
The primary aim of this study is to compare treatment for at least 90, 180, 270, and 360 days across five currently available oral anticoagulants over a study period from 2009 to 2020. The study includes three additional aims to support conclusions from the primary aim: (Aim 2) to conduct validation of key study variables and subgroup analyses within a subset of patients for whom claims and electronic health record (EHR) data are available; (Aim 3) to examine heterogeneity in treatment effects across multiple patient factors; and (Aim 4) to calibrate study results to those of available randomized trials.
Data collection & analysis
Our study will include fee-for-service Medicare claims data from 2009 to 2019 and Truven MarketScan commercial and Medicaid data from 2009 to 2020. To account for the nonrandom allocation of patients to exposure groups in our study, we will construct and apply propensity scores that summarize all potential confounders to Cox proportional hazards outcome regression models. In Aim 2, we will use EHR data to validate and refine the cohort identification definition for DVT and PE; assess and correct for residual confounding in the main claim-based analyses; incorporate the International Normalized Ratio data for warfarin patients; and examine the frequency and patterns of data missingness. In Aim 3, we will evaluate treatment effect heterogeneity with predefined subgroups and by stratifying on disease risk scores for each outcome of interest. In Aim 4, we will standardize our study results to characteristics of relevant randomized trials.
Discussion
As our population includes all Medicare novel anticoagulant users and large numbers of commercially insured and Medicaid patients, our results are likely transportable to the majority of US patients discharged from the hospital after a DVT or PE. Large, well-conducted, observational studies such as this one are particularly well suited to study rare safety events, which randomized trials may not be able to study across all comparator treatments. Results from this study will have an immediate and important impact on treatment decisions in clinical practice.
Financial & competing interests disclosure
Research reported in this publication was funded through a Patient-Centered Outcomes Research Institute (PCORI) Award (NOACs-1511-33068). The statements in this publication are solely the responsibility of the authors and do not necessarily represent the views of the Patient-Centered Outcomes Research Institute (PCORI), its Board of Governors or Methodology Committee. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as: • of interest
1.
Heit JA, Silverstein MD, Mohr DN, Petterson TM, O'Fallon WM, Melton LJ. Predictors of survival after deep vein thrombosis and pulmonary embolism: a population-based, cohort study. Arch. Intern. Med. 159(5), 445–453 (1999).
• Clinical background on deep vein thrombosis (DVT) and pulmonary embolism (PE).
2.
White RH. The epidemiology of venous thromboembolism. Circulation 107(23 Suppl. 1), I4–I8 (2003).
• Clinical background on venous thromboembolism.
3.
Schulman S, Kearon C, Kakkar AK et al. Extended use of dabigatran, warfarin or placebo in venous thromboembolism. N. Engl. J. Med. 368(8), 709–718 (2013).
• Randomized trial results comparing dabigatran to warfarin.
4.
EINSTEIN Investigators, Bauersachs R, Berkowitz SD, Brenner B et al. Oral rivaroxaban for symptomatic venous thromboembolism. N. Engl. J. Med. 363(26), 2499–2510 (2010).
• Randomized trial results comparing rivaroxaban to warfarin.
5.
Weitz JI, Lensing AWA, Prins MH et al. Rivaroxaban or aspirin for extended treatment of venous thromboembolism. N. Engl. J. Med. 376(13), 1211–1222 (2017).
• Randomized trial results comparing rivaroxaban to aspirin for extended treatment of venous thromboembolism.
6.
Agnelli G, Buller HR, Cohen A et al. Apixaban for extended treatment of venous thromboembolism. N. Engl. J. Med. 368(8), 699–708 (2013).
• Randomized trial results comparing apixaban to placebo.
7.
Kearon C, Akl EA, Comerota AJ et al. Antithrombotic therapy for VTE disease: antithrombotic therapy and prevention of thrombosis, 9th Ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 141(2 Suppl.), E419S–E496S (2012).
8.
Kearon C, Akl EA, Ornelas J et al. Antithrombotic therapy for VTE disease: CHEST guideline and expert panel report. Chest 149(2), 315–352 (2016).
9.
Savaysa (edoxaban), prescribing information. Daiichi Sankyo, Inc., Basking Ridge, NJ, USA (2015). http://dsi.com/prescribing-information-portlet/getPIContent?productName=Savaysa&inline=true.
10.
Xarelto (rivaroxaban), prescribing information. Janssen Pharmaceuticals, Inc., Titusville, NJ, USA (2015). www.xarelto-us.com/shared/product/xarelto/prescribing-information.pdf.
11.
Pradaxa® (dabigatran etexilate mesylate), prescribing information. Boehringer Ingelheim Pharmaceuticals, Inc., Ridgefield, CT, USA (2015). http://docs.boehringer-ingelheim.com/PrescribingInformation/PIs/Pradaxa/Pradaxa.pdf.
12.
Eliquis® (apixaban), prescribing information. Bristol-Myers Squibb Company and Pfizer Inc., Princeton, NJ, USA and New York, NY, USA (2015). http://packageinserts.bms.com/pi/pi_eliquis.pdf.
13.
Gagne JJ, Glynn RJ, Avorn J, Levin R, Schneeweiss S. A combined comorbidity score predicted mortality in elderly patients better than existing scores. J. Clin. Epidemiol. 64(7), 749–759 (2011).
14.
Stürmer T, Schneeweiss S, Avorn J, Glynn RJ. Adjusting effect estimates for unmeasured confounding with validation data using propensity score calibration. Am. J. Epidemiol. 162(3), 279–289 (2005).
15.
Li L, Greene T. A weighting analogue to pair matching in propensity score analysis. Int. J. Biostat. 9(2), 215–234 (2013).
16.
Yoshida K, Hernández-Díaz S, Solomon DH et al. Matching weights to simultaneously compare three treatment groups: comparison to three-way matching. Epidemiology 28(3), 387–395 (2017).
17.
Garbe E, Kloss S, Suling M, Pigeot I, Schneeweiss S. High-dimensional versus conventional propensity scores in a comparative effectiveness study of coxibs and reduced upper gastrointestinal complications. Eur. J. Clin. Pharmacol. 69(3), 549–557 (2013).
18.
Rassen JA, Schneeweiss S. Using high-dimensional propensity scores to automate confounding control in a distributed medical product safety surveillance system. Pharmacoepidemiol. Drug Saf. 21(Suppl. 1), 41–49 (2012).
19.
Schneeweiss S, Rassen JA, Glynn RJ, Avorn J, Mogun H, Brookhart MA. High-dimensional propensity score adjustment in studies of treatment effects using health care claims data. Epidemiol. Camb. Mass. 20(4), 512–522 (2009).
20.
Brown JS, Kulldorff M, Chan KA et al. Early detection of adverse drug events within population-based health networks: application of sequential testing methods. Pharmacoepidemiol. Drug Saf. 16(12), 1275–1284 (2007).
21.
Brown JS, Kulldorff M, Petronis KR et al. Early adverse drug event signal detection within population-based health networks using sequential methods: key methodologic considerations. Pharmacoepidemiol. Drug Saf. 18(3), 226–234 (2009).
22.
Silva IR, Kulldorff M. Continuous versus group sequential analysis for post-market drug and vaccine safety surveillance. Biometrics 71(3), 851–858 (2015).
23.
Li L, Kulldorff M. A conditional maximized sequential probability ratio test for pharmacovigilance. Stat. Med. 29(2), 284–295 (2010).
24.
Maro JC, Brown JS, Dal Pan GJ, Kulldorff M. Minimizing signal detection time in postmarket sequential analysis: balancing positive predictive value and sensitivity. Pharmacoepidemiol. Drug Saf. 23(8), 839–848 (2014).
25.
Seeger JD, West WA, Fife D, Noel GJ, Johnson LN, Walker AM. Achilles tendon rupture and its association with fluoroquinolone antibiotics and other potential risk factors in a managed care population. Pharmacoepidemiol. Drug Saf. 15(11), 784–792 (2006).
26.
Breiman L, Freidman J, Olshen R, Stone C. Classification and Regression Trees. Wadsworth, Belmont, CA, USA (1983).
27.
Glynn RJ, Gagne JJ, Schneeweiss S. Role of disease risk scores in comparative effectiveness research with emerging therapies. Pharmacoepidemiol. Drug Saf. 21(Suppl. 2), 138–147 (2012).
28.
Brookhart MA, Schneeweiss S. Preference-based instrumental variable methods for the estimation of treatment effects: assessing validity and interpreting results. Int. J. Biostat. 3(1), Article 14 (2007).
29.
Lipsitch M, Tchetgen Tchetgen E, Cohen T. Negative controls: a tool for detecting confounding and bias in observational studies. Epidemiol. Camb. Mass. 21(3), 383–388 (2010).
30.
Gagne JJ, Choudhry NK, Kesselheim AS et al. Comparative effectiveness of generic and brand-name statins on patient outcomes: a cohort study. Ann. Intern. Med. 161(6), 400–407 (2014).
31.
Lash T, Fox M, Fink A. Applying quantitative bias analysis to epidemiologic data. Springer, NY, USA (2009).
32.
Lash TL, Fox MP, MacLehose RF, Maldonado G, McCandless LC, Greenland S. Good practices for quantitative bias analysis. Int. J. Epidemiol. 43(6), 1969–1985 (2014).
33.
Tamariz L, Harkins T, Nair V. A systematic review of validated methods for identifying venous thromboembolism using administrative and claims data. Pharmacoepidemiol. Drug Saf. 21(Suppl. 1), 154–162 (2012).
34.
Wahl PM, Rodgers K, Schneeweiss S et al. Validation of claims-based diagnostic and procedure codes for cardiovascular and gastrointestinal serious adverse events in a commercially-insured population. Pharmacoepidemiol. Drug Saf. 19(6), 596–603 (2010).
35.
Birman-Deych E, Radford MJ, Nilasena DS, Gage BF. Use and effectiveness of warfarin in Medicare beneficiaries with atrial fibrillation. Stroke 37(4), 1070–1074 (2006).
Information & Authors
Information
Published In
Copyright
© 2017 Future Medicine Ltd.
History
Received: 14 July 2017
Accepted: 22 September 2017
Published online: 21 December 2017
Keywords:
Topics
Authors
Metrics & Citations
Metrics
Article Usage
Article usage data only available from February 2023. Historical article usage data, showing the number of article downloads, is available upon request.
Citations
How to Cite
Study protocol for the dabigatran, apixaban, rivaroxaban, edoxaban, warfarin comparative effectiveness research study. (2017) Journal of Comparative Effectiveness Research. DOI: 10.2217/cer-2017-0053
Export citation
Select the citation format you wish to export for this article or chapter.