Beyond external control arms: Leveraging real-world data to support regulatory applications in oncology

Randomized controlled trials (RCTs) are the gold standard for clinical research; however, in many areas, like oncology, they are infeasible because new therapies often target rare tumors and, therefore, the indicated population is small. Single-arm trials are frequently used as a feasible alternative to evaluate a treatment's effectiveness without a traditional control group. The interpretation of the results from a single-arm trial can be supported by a real-world external control arm (ECA)—where real-world data (RWD) are used to estimate the trial outcome rate in a group of patients who have not received the investigational treatment for comparison with the outcome rate observed in the single-arm trial. ECAs have been used successfully to support FDA approval of rare disease therapies and are increasingly being explored in clinical development. In oncology applications, however, FDA reviewers have found their utility to be limited. Given this, can RWD be useful in supporting oncology regulatory applications, beyond the use in ECAs?
During the 2024 Drug Information Association Real-World Evidence (DIA RWE) Conference (October 24–25, 2024, Philadelphia, PA, USA), Ulka Campbell (Head of Scientific Strategy, Aetion, Inc., USA) explored this topic in the presentation, “Beyond External Control Arms: Utility of Real-World Data in Oncology Regulatory Applications.” Here, we take a Deep Dive into the session, summarizing the key discussions and main takeaways from the oncology case studies illustrating how RWD can support regulatory decision-making, such as rationalizing trial design and assuring the representativeness of trial populations.
Regulatory acceptability of ECAs
Campbell began the presentation by providing an overview of the applicability of oncology ECAs by FDA reviewers. ECAs submitted to support effectiveness claims have generally not been accepted by the FDA, largely due to differences in data collection between trials and routine clinical practice. Campbell noted that in oncology approvals, single-arm trials are often deemed sufficient for meeting clinical effectiveness evidence standards, making the added value of ECAs uncertain in this field. This contrasts with approvals outside of oncology, where ECAs have been accepted as part of adequate and well-controlled investigations necessary for approval.
Other use cases of RWD in oncology applications
After outlining the limited success of ECAs for regulatory decision-making, Campbell shifted focus to the broader utility of RWD in regulatory applications. She highlighted how RWD have been valuable in oncology drug approvals in other ways, beyond ECAs.
The team at Aetion conducted a systematic review of 76 publicly available CBER and CDER approvals of oncology therapies from 2019 to 2023. Using a structured extraction form, they captured information, including the type of RWD (e.g., previously published studies or new analyses by sponsors) and its purpose (supporting safety, effectiveness, or providing contextual evidence). Additionally, the review noted how FDA reviews considered the RWD—whether it contributed substantial or supportive evidence of effectiveness, or helped rationalize trial design, interpret findings, or characterize the disease condition or current therapy options.
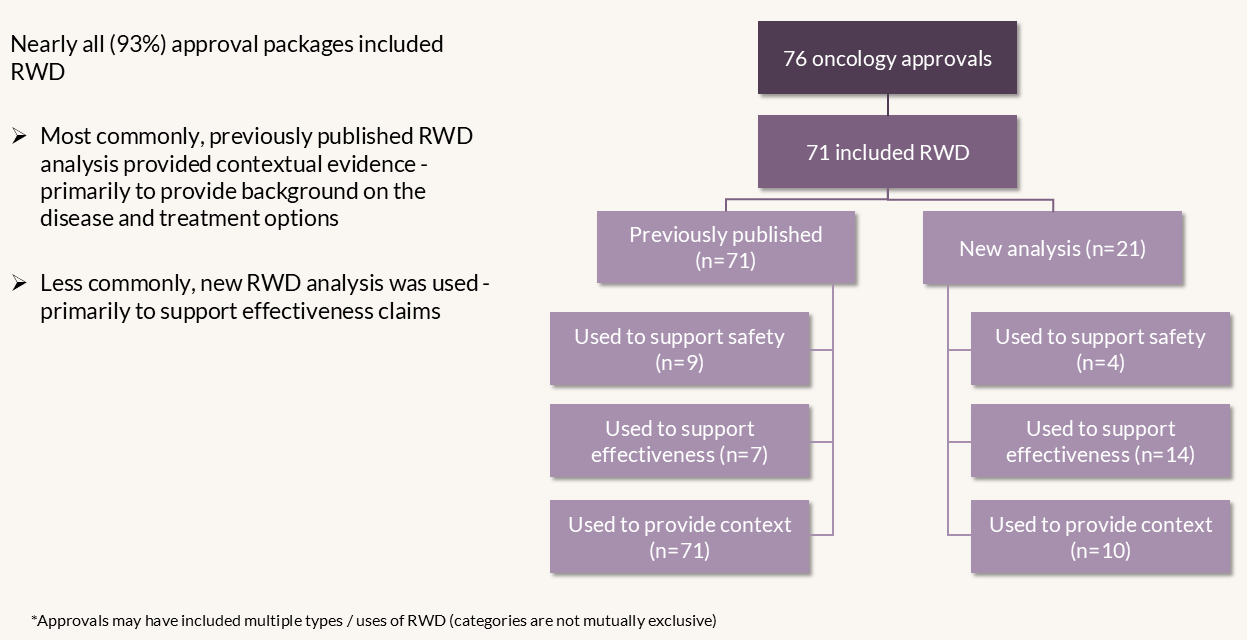
The analysis found that, of the 76 oncology approvals reviewed, nearly all (n=71) included RWD. All these, 71 approvals included previously published RWD studies; 7 approvals included published RWD to support evidence of effectiveness, and 9 approvals included published RWD to support evidence of safety. In 21 approvals, sponsors provided new RWD analyses—most (n=14) to support effectiveness claims, with 4 focused on safety and 10 providing context. Of the 14 use cases that involved new analysis of RWD to support effectiveness, 7 were ECAs. Campbell noted the main concerns raised by FDA reviewers related to ECAs, including: immortal time bias, differing inclusion/exclusion criteria, inadequate control of confounding factors, treatment misclassification, biased interpretation of outcomes (e.g., lead time bias, incomplete death data), and small sample size.
Campbell next presented six case studies selected from the analysis to illustrate the value of RWD in supporting oncology regulatory applications.
Case studies
The first two case studies illustrated the use of previously published RWD to support FDA’s interpretation of pivotal single-arm trial results.
Case study 1: Previously published RWD analysis demonstrates lack of spontaneous remission of disease
For ropeginterferon alfa-2b-NJFT (Besremi®), a treatment for polycythemia vera, published natural history data showed that spontaneous remission of the disease is rare and highlighted the significant risks of thrombosis and death when untreated. These RWD were cited as supporting FDA’s finding of substantial evidence of effectiveness.
Key takeaway: Published real-world studies can be valuable in regulatory applications beyond routine characterization of the disease condition.
Case study 2: Previously published RWD analysis supports endpoint threshold and size of pivotal trial
For belantamab mafodotin-blmf (Blenrep®), developed to treat relapsed, refractory multiple myeloma, the sponsor used published RWD and other sources to create an outcome rate benchmark to inform the design of their pivotal single-arm trial. Specifically, an objective response rate (ORR) threshold of 15% was used to calculate the sample size requirement for the trial. The trial met its primary endpoint, defined as the lower limit of the confidence interval for the observed ORR exceeding the 15% threshold. FDA reviewers noted the trial design rationale and that the ORR findings provided substantial evidence of effectiveness.
Key takeaway: RWD can be intentionally used to design clinically relevant trials.
Campbell next outlined four case studies demonstrating the value of new RWD analysis for supportive context.
Case study 3: New RWD analysis demonstrates unmet need
To support the application for capmatinib (Tabrecta®) for advanced MET-mutated non-small cell lung cancer (NSCLC), a global chart review study was conducted to provide key insights into the natural history of the diseases. The FDA found that the natural history study provided clinically significant information that the treatment addresses an unmet medical need, and, relatedly, that the MET mutation is a negative prognostic factor predicting poor response to standard therapies. Notably, this study was discussed in an end-of-Phase 2 meeting, where the FDA emphasized that, alongside evidence of treatment benefit, the natural history study findings would be considered in the application review.
Key takeaway: Early FDA engagement on a natural history study is valuable for understanding its potential role in regulatory application, even if it is not part of the pivotal evidence strategy.
Case study 4: New RWD analysis provides benchmarks for assessing representativeness of trial population
CDER Multi-disciplinary Review and Evaluation
In the approval of tisotumab vedotin-tftv (Tivdak®) for advanced cervical cancer, it was noted that only 15% of the pivotal trial participants were from the US, with the remainder recruited from the European Union. To mitigate concerns about the generalizability of trial findings to the US population, an analysis of RWD from health claims and SEER (Surveillance, Epidemiology, and End Results) data was conducted to characterize treatment utilization and disease factors among US patients with cervical cancer. Based on this analysis, the FDA concluded that the trial population was reflective of US patients.
Key takeaway: Comprehensive understanding of the baseline characteristics of the indicated population is essential for assessing representativeness of the trial population and generalizability of findings to the intended patient population.
Case study 5: New RWD analysis informs pivotal trial sample size
In the approval of omidubicel-onlv (Omisirge®) for patients undergoing umbilical cord blood transplantation for hematologic malignancies, RWD from the Center for International Blood and Marrow Transplant Research (CIBMTR) registry provided information on the trial endpoint in untreated patients needed for the pivotal trial sample size calculation.
Key takeaway: Robust analysis of RWD can provide critical information needed to design pivotal trials, such as determining sample size.
Case study 6: New RWD analysis supports the rationale for a non-randomized pivotal trial
In the final case study, Campbell discussed the use of RWD to rationalize the lack of randomization and control group in the pivotal trial (which was a single-arm trial) for idecabtagene vicleucel (Abecma®), a treatment for relapsed, refractory multiple myeloma. The sponsor combined RWD from registries, clinical sites, and other external research databases to create an ECA. Although the FDA did not find the ECA acceptable for direct comparison with the single-arm trial, it acknowledged that given the wide variability in real-world treatments and the lack of an established standard of care observed within the RWD, a randomized pivotal trial was infeasible and concluded that a single-arm trial was justified.
Key takeaway: Deep and comprehensive understanding of treatment utilization and standard of care within the indicated population is important to showing that a randomized pivotal trial design may be infeasible.
Lessons learned for integrated real-world evidence generation
For oncology treatments, the inherent differences in data collection between clinical trials and routine practice make direct comparisons between single-arm trials and ECAs difficult to interpret and challenging for regulatory decisions. However, a review of FDA approval use cases shows how RWD can be more effectively utilized to support oncology clinical development, beyond the use of ECAs.
Campbell emphasized that for a truly integrated real-world evidence-generation approach, early incorporation of real-world studies into the clinical development plan is essential. This process should begin with a comprehensive literature review of the baseline factors, treatment utilization, and natural history of the specific indicated population. Following that, robust RWD analysis should be conducted to address gaps in the literature and provide current information on the indicated population. By establishing a real-world study infrastructure early, and periodically updating the analyses, researchers will have necessary and timely information to support critical decisions throughout development, such as pivotal trial design.
“If you have a comprehensive knowledge of your target population through RWD, you can use that insight to inform trial design and endpoint definitions, ensuring a more 'apples-to-apples' comparison with benchmarks. Having a deep understanding of the indicated population as they are treated in real-world care is critically important to setting yourself up for successful and interpretable results.”
While the presentation focused on oncology examples, Campbell emphasized that the use of RWD is critical for clinical development of all therapeutic areas.

Sponsorship for this Deep Dive was provided by Aetion